

The emphasis on pressure cascade and airflow protection is not a new concept. In fact, earlier versions of EU Annex 1, dating back to the 1990s and early 2000s, required positive pressure differentials between cleanroom grades, but the requirement was worded more generally. Facilities were expected to maintain pressure differentials “as appropriate,” which left room for interpretation. As a result, applications varied widely between manufacturers. Some companies set targets of 5 pascals, while others maintained higher differentials; in some cases, monitoring was limited to periodic manual checks rather than continuous measurement [1].
Over the years, regulators have observed that the inconsistent implementation of pressure cascades contributes to contamination risks. Several inspection findings in Europe highlighted cases where inadequate pressure control allowed for potential cross-contamination between Grade B and C areas or compromised background conditions that supported Grade A environments. These events provided the evidence base for tighter harmonization.
 The 2022 revision of Annex 1 sharpened expectations by codifying a minimum 10 pascal differential between adjacent grades (Clause 4.14), [1]. Unlike earlier versions, the requirement is now unambiguous, enforceable, and backed by an expectation of continuous monitoring with alarms, rather than relying on static qualification data.
The 2022 revision of Annex 1 sharpened expectations by codifying a minimum 10 pascal differential between adjacent grades (Clause 4.14), [1]. Unlike earlier versions, the requirement is now unambiguous, enforceable, and backed by an expectation of continuous monitoring with alarms, rather than relying on static qualification data.
This revision also reflects a broader shift in regulatory philosophy: structural requirements, such as fixed differentials, are essential, but they are complemented by performance-based verification. Annex 1, therefore, integrates pressure cascade rules with broader contamination control strategy (CCS) principles, ensuring that airflow integrity, smoke visualization, and environmental monitoring all reinforce each other.
Globally, regulators have taken slightly different approaches. The Pharmaceutical Inspection Co-operation Scheme (PIC/S), which aligns inspection standards across more than 50 countries, mirrors the EU model almost directly, emphasizing pressure cascades and continuous monitoring as a fundamental principle. The World Health Organization (WHO), particularly in its guidance for vaccine and parenteral production, also closely mirrors Annex 1, reinforcing cleanroom zoning and differential pressures as part of aseptic assurance [1].
By contrast, Japan’s PMDA and the USFDA have historically focused less on static numbers and more on airflow visualization and contamination risk management. The FDA’s aseptic guidance [2] emphasizes airflow velocity, smoke studies, and environmental monitoring data as the primary indicators of protection for Grade A zones. Japan’s regulators have long required firms to show through airflow mapping and EM data that critical zones remain protected under dynamic conditions.
The trend across all regulators is convergence. Europe codifies structural differentials, while the US and Japan stress performance-based demonstrations. However, both perspectives acknowledge that airflow dynamics and environmental monitoring data ultimately serve as the decisive proof of sterility assurance. The harmonization is not complete, but manufacturers today are expected to design systems that satisfy both philosophies: robust cascades across closed boundaries and evidence-driven airflow protection in open zones.
Designing and maintaining a qualified pressure cascade is not without engineering challenges. Achieving and consistently holding ≥10 pascals across cleanroom boundaries requires far more than setting fan speeds – it demands precise HVAC zoning, accurate balancing of supply and exhaust airflows, and robust architectural integrity. Even minor weaknesses, such as leaks around door seals, poorly sealed pass-throughs, ceiling penetrations, or floor drains, can destabilize differentials and compromise contamination control. Pressure fluctuations may also occur due to external factors, such as weather-driven changes in building pressurization or sudden door openings during peak operations [1][3].
To counteract these risks, modern cleanroom facilities employ a combination of engineering redundancies and structural safeguards. Airlocks serve as controlled buffer spaces, reducing the risk of pressure collapse when personnel or materials move between grades. Pressure stabilizers maintain steady airflow even when HVAC loads fluctuate, while door interlocking systems prevent simultaneous openings that could rapidly equalize pressures between rooms. In advanced installations, these interlocks are integrated into Building Management Systems (BMS), ensuring automated fail-safes in the event of any deviation that threatens to compromise cascade stability.
 HVAC zoning design plays a crucial role in achieving optimal cascade performance. Each cleanroom grade is supplied with independent supply and return ducts, preventing backflow or cross-contamination between areas of different cleanliness. Supply air is typically HEPA-filtered at terminal points to guarantee quality, while return air paths are strategically located to create sweeping airflow patterns that reinforce unidirectionality. In high-risk operations – such as sterile filling or cytotoxic compounding – pressure relief dampers are installed to allow controlled air release without compromising the cascade, thereby protecting both product and operators.
HVAC zoning design plays a crucial role in achieving optimal cascade performance. Each cleanroom grade is supplied with independent supply and return ducts, preventing backflow or cross-contamination between areas of different cleanliness. Supply air is typically HEPA-filtered at terminal points to guarantee quality, while return air paths are strategically located to create sweeping airflow patterns that reinforce unidirectionality. In high-risk operations – such as sterile filling or cytotoxic compounding – pressure relief dampers are installed to allow controlled air release without compromising the cascade, thereby protecting both product and operators.
Another challenge lies in maintaining stability under dynamic operating conditions. While differential pressure may hold during qualification testing, actual operations introduce variables such as personnel movement, door cycling, and equipment heat loads. Regulators expect firms to demonstrate that differentials are not only established during commissioning but also sustained during worst-case operational scenarios [1]. This requires integrating engineering controls with ongoing monitoring and alarm systems, ensuring that deviations are detected in real-time and investigated within the Quality Management System (QMS).
Ultimately, these technical measures transform Annex 1’s structural requirement for a 10–15 Pa pressure differential from a theoretical target into a practical, sustainable safeguard. By combining robust facility design, precise HVAC balancing, and intelligent automation, manufacturers can demonstrate to regulators that cleanroom cascades remain reliable under the complex and variable realities of aseptic operations [1][3].
Pressure and airflow must be demonstrated not just at the time of facility qualification but also continuously throughout operation. Regulators now expect companies to prove that their contamination control measures are effective under dynamic conditions, not just during static tests. For instance, airflow velocity in Grade A laminar airflow (LAF) units is typically measured at commissioning to confirm compliance with the expected benchmark of 0.45 m/s ± 20%. Still, this parameter must also be re-verified at least annually or whenever significant changes occur, such as major HVAC modifications, filter replacements, or equipment reconfigurations [2][3].
Airflow visualization studies (smoke studies) are equally critical. These studies are first conducted during qualification to demonstrate that unidirectional airflow effectively sweeps contaminants away from critical product exposure zones. However, Annex 1 (2022) and FDA inspectors increasingly emphasize that smoke studies must also reflect worst-case operational scenarios – including operator interventions, equipment setup, and dynamic activities. Facilities are therefore expected to repeat these studies not only after significant modifications but also periodically, often every one to two years, to confirm ongoing airflow integrity [2][3].
Continuous differential pressure monitoring at sealed cleanroom boundaries has become a standard of good practice. Sensors integrated into Building Management Systems (BMS) provide real-time visibility of differential pressure trends, trigger alarms for excursions, and enable automated recording. By combining this monitoring into the Quality Management System (QMS), deviations are thoroughly investigated, documented, and resolved with corrective and preventive actions (CAPA). This integration also generates a strong audit trail, which inspectors view as evidence that the facility is operating in a state of control rather than reacting to problems retrospectively.
Nevertheless, monitoring technologies have inherent limitations. Non-viable particle counters are excellent for measuring airborne particulate cleanliness in real time, but they cannot distinguish between inert particles and viable microorganisms. Microbiological monitoring – using settle plates, active air samplers, and contact plates – remains indispensable. However, these methods suffer from a time lag due to incubation, meaning contamination is only confirmed after operations are complete. This lag leaves facilities reliant on a multi-layered monitoring program combined with risk-based interpretation of data to manage contamination risks effectively [3][4]. As regulators often highlight, genuine sterility assurance is built not on a single monitoring tool but on the convergence of multiple independent controls.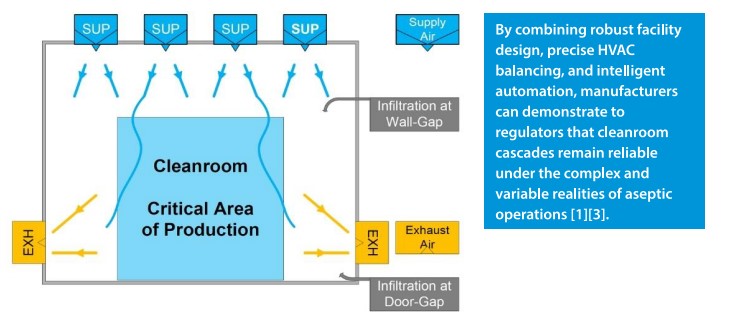
The future of cleanroom monitoring is shifting toward real-time, data-rich control strategies. Emerging technologies such as AI-driven analytics and machine learning models promise to identify subtle patterns in airflow or contamination data long before they become deviations. Novel real-time viable particle sensors are under development, offering the possibility of continuous microbiological monitoring rather than delayed plate-based methods. Computational Fluid Dynamics (CFD) modelling is also becoming more widespread, allowing facilities to simulate airflow interactions under dynamic conditions and optimize designs before construction.
Unlike static qualification tests, CFD provides a three-dimensional, time-resolved visualization of how air moves within cleanrooms, isolators, and laminar flow hoods. Engineers can model variables such as air velocity, turbulence, particle transport, heat loads from equipment, and the impact of operator movement. This predictive analysis helps identify dead zones, recirculation pockets, or turbulence that could compromise unidirectional airflow long before the room is built or commissioned.
For example, CFD can reveal whether a planned HVAC diffuser layout will generate sufficient sweeping of particles away from Grade A work zones or whether operator interventions may disturb airflow patterns. The results allow designers to adjust air supply positions, exhaust locations, and pressure balancing strategies during the design stage, rather than relying solely on physical smoke studies after construction. While smoke visualization remains essential for regulatory demonstration, CFD complements it by offering a proactive, risk-based tool that reduces costly design changes post-installation. Increasingly, regulators view CFD data as valuable supporting evidence during inspections, especially when firms can demonstrate how modelling outcomes informed design decisions that align with Annex 1 contamination control strategy expectations [1][2].
Looking forward, regulators may continue to refine their guidance. Future revisions of Annex 1 or FDA aseptic guidancemay place greater emphasis on performance-based data, aligning structural requirements, such as the 10 pascal rule, with dynamic verification tools, including smoke studies and continuous monitoring. The key question is whether pressure cascade will remain a cornerstone requirement, or whether performance-driven evidence will eventually take precedence. For now, the most robust compliance strategy is one that embraces both structural cascade controls for closed boundaries and dynamic airflow-based evidence for open zones.
- EU GMP Annex 1: Manufacture of Sterile Medicinal Products. European Commission, 2022.

- USFDA Guidance for Industry: Sterile Drug Products Produced by Aseptic Processing – Current Good Manufacturing Practice. U.S. Food and Drug Administration, 2004.
- Sandle, T. Cleanroom Management in Pharmaceuticals and Healthcare. Woodhead Publishing, 2016.
- Whyte, W. Cleanroom Technology: Fundamentals of Design, Testing and Operation. Wiley, 2010.
 About the Author
About the Author
Dr. Tarun Chugh is a seasoned pharmaceutical professional with over 34 years of extensive experience in Microbiology Quality Control and Quality Assurance. Currently serving as the CEO of SIMco Pharma Consultancy, based in Ahmedabad, Dr. Chugh specializes in conducting gap analyses for manufacturing units in various sectors, including Formulations (OSD/Sterile), API (Sterile/Non-Sterile), Excipients, and Packaging Materials. His expertise spans a wide range of regulatory standards such as USFDA, MHRA, EU, PICs, TGA, ANVISA, and WHO.
Dr. Chugh is a recognized authority in Quality Systems, Microbiology techniques, and various forms of validation, including Clean Room, Aseptic, Process, Water Systems, and HVAC. He is adept at handling Quality Management Systems (QMS), performing risk assessments, and ensuring data integrity (ALCOA+). Additionally, he is skilled in technical transfers, troubleshooting, and implementing remedial actions. Dr. Chugh is also highly regarded for his ability to provide specialized training in Quality Systems, Microbiology, and Aseptic behaviors, making him a valuable resource in the pharmaceutical industry.