


 By Dr. Binh Nguyen
By Dr. Binh Nguyen
Dr. Binh Nguyen, a retired USFDA officer and founder and CEO of Wynngate Consulting, discusses ongoing quality issues in the bio/pharmaceutical industry and offers solutions aimed at advancing quality and stabilizing the drug supply chain to ensure the delivery of high-quality products to patients. The focus of this article is on US FDA market and is broken into 3 sections: compliance data, FDA’s response to quality issues, and author’s quality solutions.
DID YOU KNOW? Drugs and biologics have the highest (68.5%) import actions (Table 1) among all FDA regulated commodities in 2023? Per 2023 FDA Import Data (Figure 1), only 2% of drugs and biologics are imported into the USA among other FDA regulated commodities.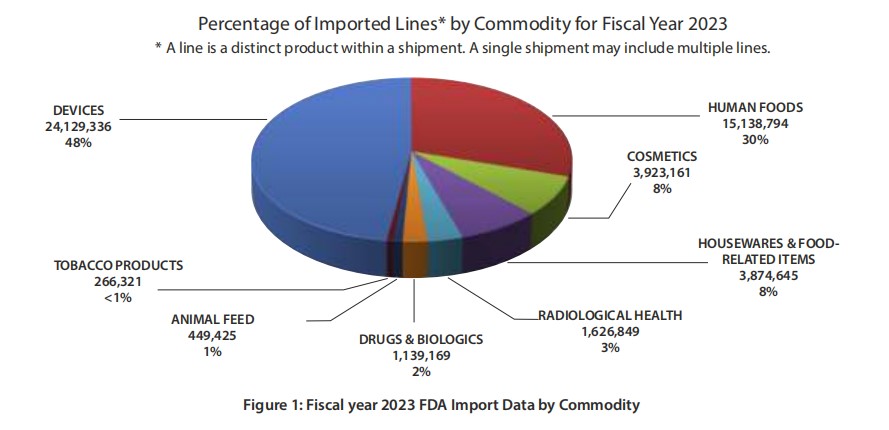
Yet, in fiscal year 2023, out of 14,417 refusals of all commodities at import, 3,290 (22.8%) of drugs and biologics were being refused at import (see Figure 2).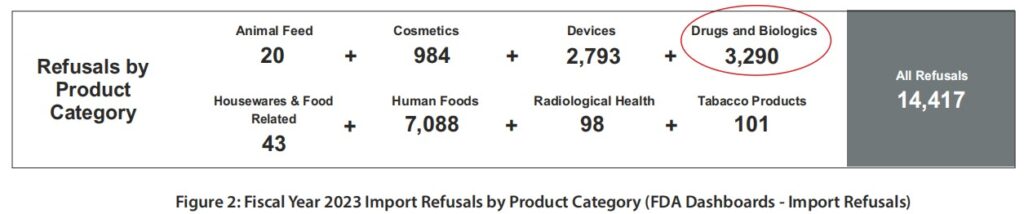
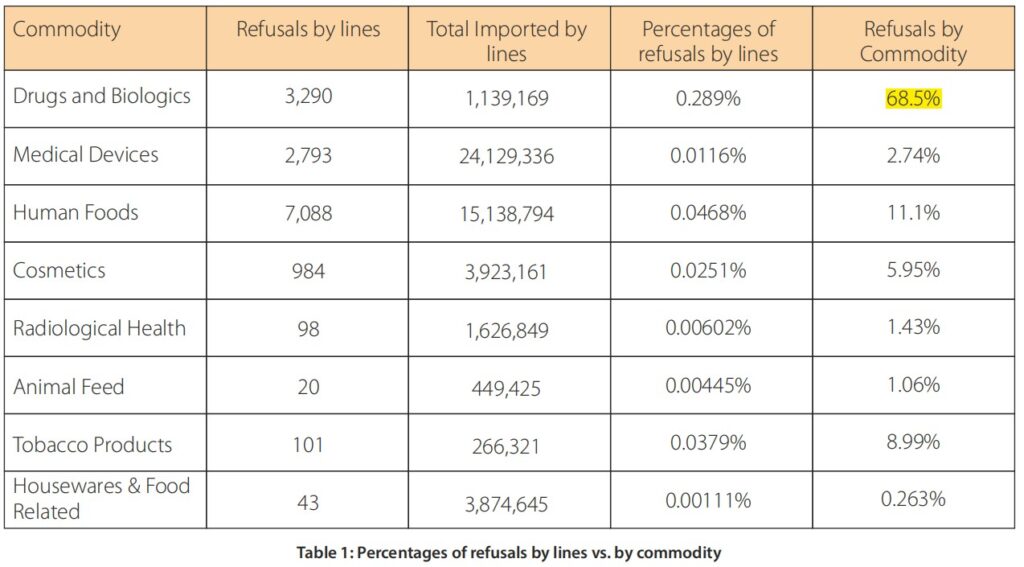
Based on the number of refusals by product category in 2023, here are the refusal percentages by imported lines and by commodity (see Table 1).
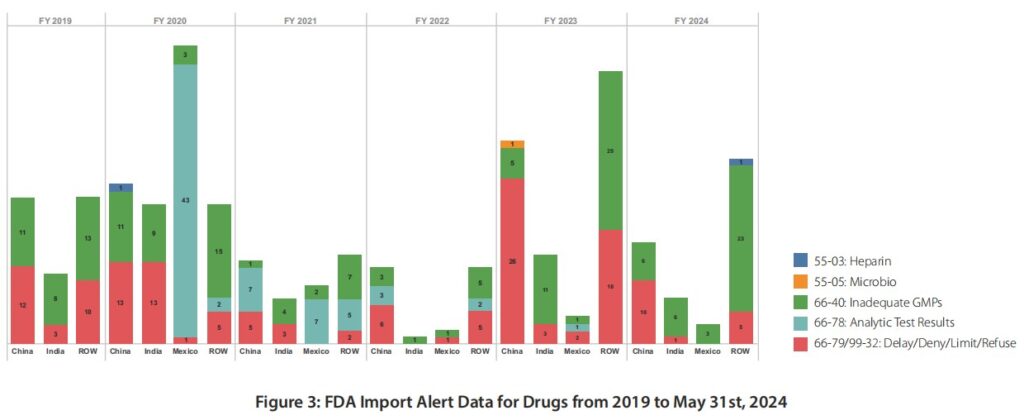
 What are the reasons for these refusals at Import? Per FDA Import Alert Data from 2019 to May 31, 2024 (Figure 3), inadequate CGMPs (>44%) take precedence over those years compared to other deficiencies although Mexico had the highest rate of refusals based on analytical test results in 2020 during COVID-19 and China had the highest rate of delaying/denying/limiting/refusing inspections in 2023.
What are the reasons for these refusals at Import? Per FDA Import Alert Data from 2019 to May 31, 2024 (Figure 3), inadequate CGMPs (>44%) take precedence over those years compared to other deficiencies although Mexico had the highest rate of refusals based on analytical test results in 2020 during COVID-19 and China had the highest rate of delaying/denying/limiting/refusing inspections in 2023.
Note: due to COVID-19 in 2020, the number of onsite inspections had reduced significantly. It is worth to note that in 2023, there is one Import Alert 55.05 for “Detention without Physical Examination of Finished Dosage Form Drug Products, Active Pharmaceutical Ingredients, and Inactive Ingredients for Potentially Hazardous Microbiological Contamination” – see Import Alert 55-05 (fda.gov). Import Alert 55.03 for “Detention without Physical Examination of Different Forms of Heparin and Heparin-Related Products” also resurfaced in 2024 from 2020 – see Import Alert 55-03 (fda.gov).
 A deeper dive into FY2023 Drug-Quality-Related Import Alerts (see Figure 4) from Fiscal Year 2023 Report on the State of Pharmaceutical Quality (fda.gov) shows that India had the highest number of Import Alerts by GMP-based inspections, South Korea had the highest Import Alerts by GMP-based 704(a)(4), and China had the highest Import Alerts by refusal-based 704(a)(4) record request.
A deeper dive into FY2023 Drug-Quality-Related Import Alerts (see Figure 4) from Fiscal Year 2023 Report on the State of Pharmaceutical Quality (fda.gov) shows that India had the highest number of Import Alerts by GMP-based inspections, South Korea had the highest Import Alerts by GMP-based 704(a)(4), and China had the highest Import Alerts by refusal-based 704(a)(4) record request. 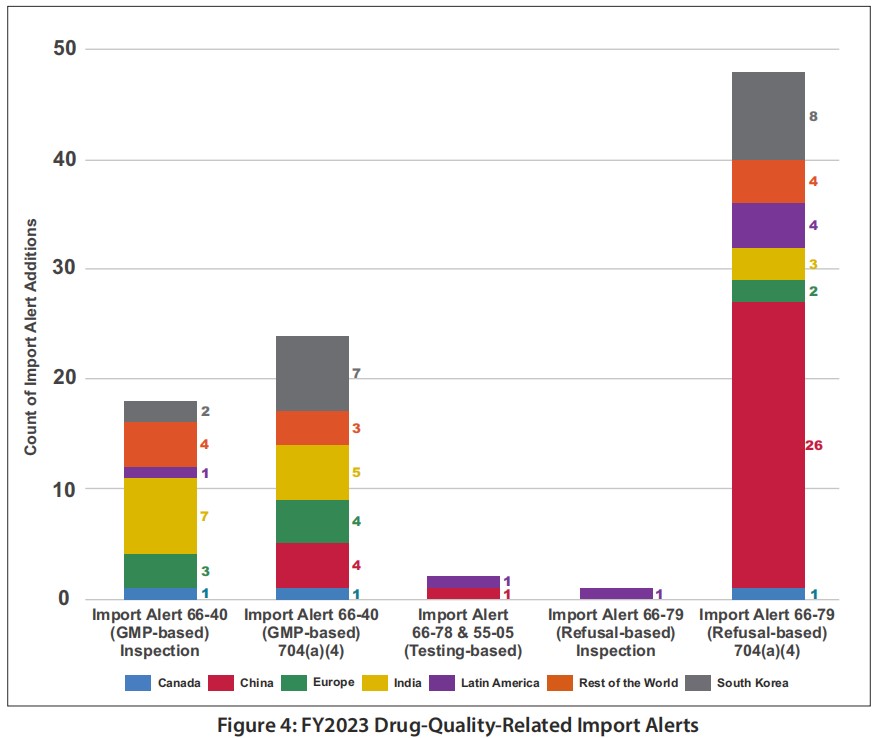 There is an increase in import alert additions related to site inspections (19 in FY2023 compared with 6 and 7, respectively, in FY2022 and FY2021). Most of the sites placed on quality-related import alert (90%) are manufacturers of OTC monograph drug products (see Figure 4 for further explanation on why this is the case).
There is an increase in import alert additions related to site inspections (19 in FY2023 compared with 6 and 7, respectively, in FY2022 and FY2021). Most of the sites placed on quality-related import alert (90%) are manufacturers of OTC monograph drug products (see Figure 4 for further explanation on why this is the case).
Besides import alert, FDA also uses other regulatory tools such as injunctions, regulatory meetings, consent decrees, seizures, warning letters, untitled letters, and administrative detention. Per FDA Office of Compliance nnual Report Fiscal Year 2023, CGMP violations accounted for the highest (45%) percentage among different types of violations (Figure 5).
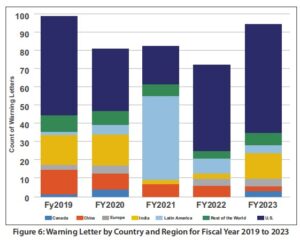
So which countries get the most warning letters for pharmaceuticals? Per Fiscal Year 2023 Report on the State of Pharmaceutical Quality (fda.gov), FDA warning letters data from 2019 to 2023, US companies still faced with the highest number of warning letters followed by fluctuating data between China and India with Mexico had a spurt in 2021 as shown in Figure 6.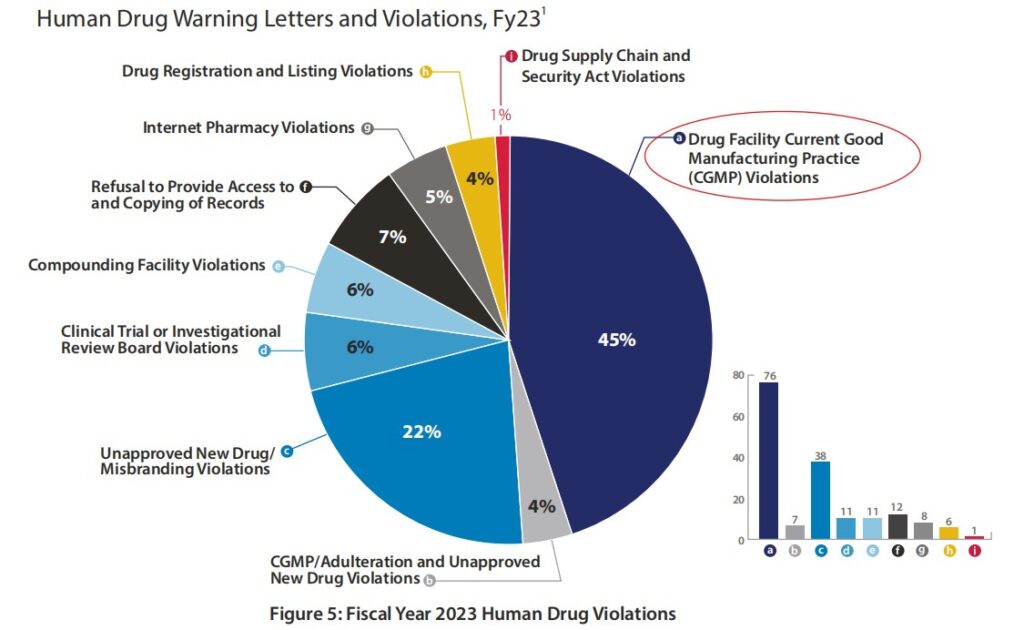
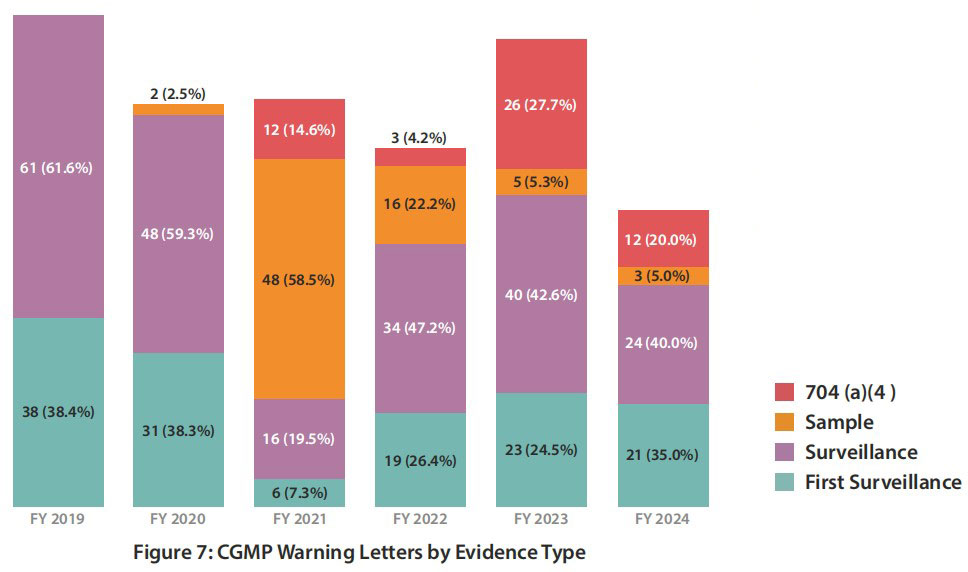
 Note: Warning letter data is based on commercially marketed products. Figure 7 shows warning letters are acted upon by FDA via surveillance inspections (or routine CGMP inspections), followed by first surveillance inspection (or first CGMP inspection), then 704(a)(4) or record requests, then lastly sampling (with the exception of 2021 where sampling had the highest percentage of warning letters due to COVID-19 limiting onsite FDA inspections). The data shown in Figure 7 from Oct 1st, 2018 to May 31st, 2024 does not include compounding pharmacies’ statistics.
Note: Warning letter data is based on commercially marketed products. Figure 7 shows warning letters are acted upon by FDA via surveillance inspections (or routine CGMP inspections), followed by first surveillance inspection (or first CGMP inspection), then 704(a)(4) or record requests, then lastly sampling (with the exception of 2021 where sampling had the highest percentage of warning letters due to COVID-19 limiting onsite FDA inspections). The data shown in Figure 7 from Oct 1st, 2018 to May 31st, 2024 does not include compounding pharmacies’ statistics.
 One may ask why bio/pharmaceutical industries face so much regulatory challenges by FDA despite many drugs (A/NDA) going through application and facility inspection prior to entering the commercial market. In reality, OTC drug manufacturers make up the majority of the warning letters issued by FDA from the same time period of Oct 1st, 2018 to May 31st, 2024 as depicted in Figure 8.
One may ask why bio/pharmaceutical industries face so much regulatory challenges by FDA despite many drugs (A/NDA) going through application and facility inspection prior to entering the commercial market. In reality, OTC drug manufacturers make up the majority of the warning letters issued by FDA from the same time period of Oct 1st, 2018 to May 31st, 2024 as depicted in Figure 8.
This makes sense as OTC drug manufacturers do not have to go through the rigorous drug approval process (unless it was a prescription drug later converted into OTC drug) with facility process inspection as part of a pre-approval inspection and one can see such facilities (Rx and OTC/Rx with pre-approval inspections) have much less (but not zero) warning letters after commercialization. In other words, OTC drug manufacturers when getting inspection visits from FDA may not as prepared to deal with CGMP inspections compared to companies that have spent much preparation to be ready for pre-approval inspections and eventually surveillance inspections.
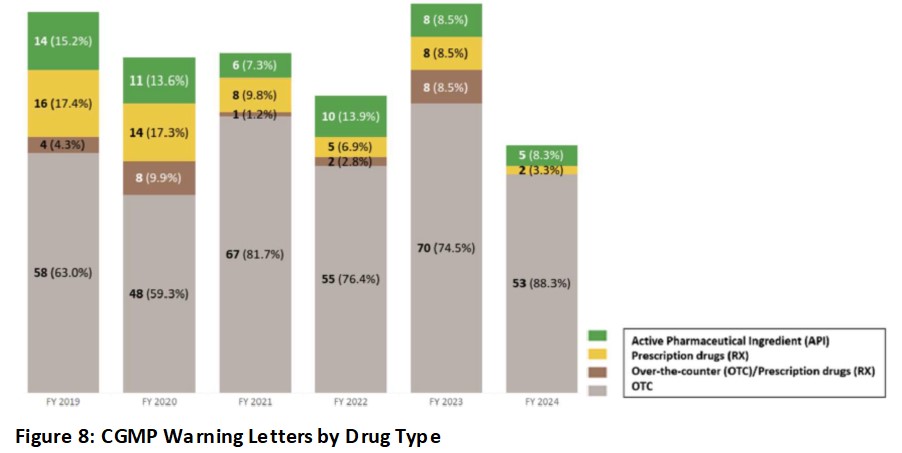 It is disheartening to continue to see drug shortages going back many decades and CGMP deficiencies continue to lead the chart as the main cause. In my 20+ years of working at the FDA, I witnessed many drug shortages and had also experienced it firsthand as a hospital pharmacist where knowledge from FDA inspections helped me understand the causes and saw the effects on patients. Drug shortages continue to be an ongoing problem today. FDA acknowledges that quality problems have been the most common reason for shortages as shown in Figure 9 for CY2022 and CY2023 per Fiscal Year 2023 Report on the State of Pharmaceutical Quality (fda.gov) (Note: increases in demand were due to COVID-19 not previously seen also accounted for similar drug shortage percentages as quality issues/manufacturing delays).
It is disheartening to continue to see drug shortages going back many decades and CGMP deficiencies continue to lead the chart as the main cause. In my 20+ years of working at the FDA, I witnessed many drug shortages and had also experienced it firsthand as a hospital pharmacist where knowledge from FDA inspections helped me understand the causes and saw the effects on patients. Drug shortages continue to be an ongoing problem today. FDA acknowledges that quality problems have been the most common reason for shortages as shown in Figure 9 for CY2022 and CY2023 per Fiscal Year 2023 Report on the State of Pharmaceutical Quality (fda.gov) (Note: increases in demand were due to COVID-19 not previously seen also accounted for similar drug shortage percentages as quality issues/manufacturing delays).
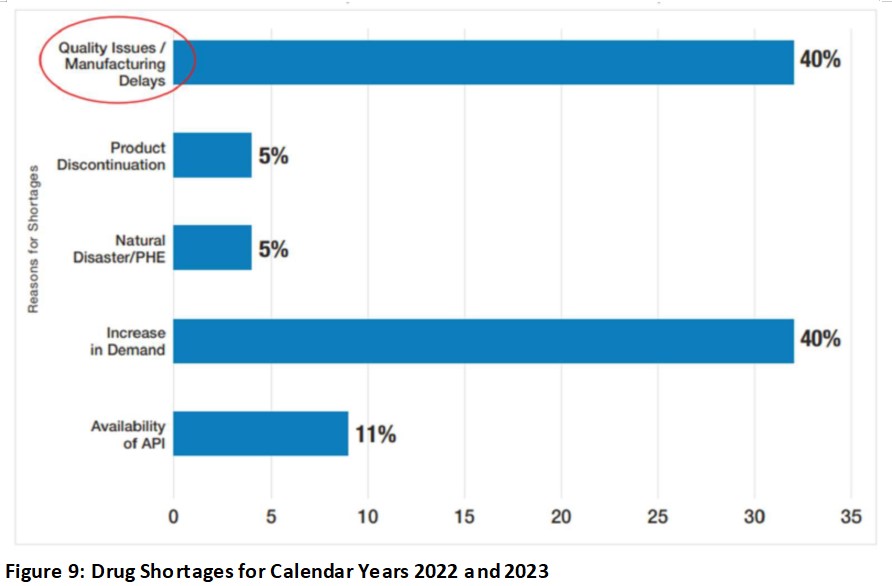
Similar to drug shortages, recalls are also topped with GMP deviations as shown in Figure 10 for FY2019 to FY2023. Note that recalls due to contamination increased in FY2023 and failed specifications also stayed relatively consistently significant from FY2019 to FY2023. In my opinion, all these types of recalls listed in Figure 10 are related to quality or CGMP issues with the exception of labelling.
 During FY2023 per Fiscal Year 2023 Report on the State of Pharmaceutical Quality (fda.gov), 35% of recalled products were associated with five specific events where two events were related to data integrity and two events were related to sterility assurance:
During FY2023 per Fiscal Year 2023 Report on the State of Pharmaceutical Quality (fda.gov), 35% of recalled products were associated with five specific events where two events were related to data integrity and two events were related to sterility assurance:
- 86 product recalls attributed to a single manufacturer that went out of business and was, therefore, unable to maintain CGMP requirements for distributed drugs, such as stability testing.
- 67 product recalls attributed to a single drug manufacturing site for data integrity issues identified during an inspection.
- 52 product recalls attributed to a single drug manufacturing site for lack of assurance of sterility.
- 17 product recalls attributed to a single manufacturing site for data integrity issues identified during an inspection.
- 17 product recalls attributed to a single manufacturing site for lack of assurance of sterility.
If you haven’t personally felt the impact of drug recalls yet, perhaps the FDA chart (Figure 11) for FY2023 may help you relate to people you may know who may take one of these types of medications. Antibiotics had the highest percentage of recalls in FY 2023.
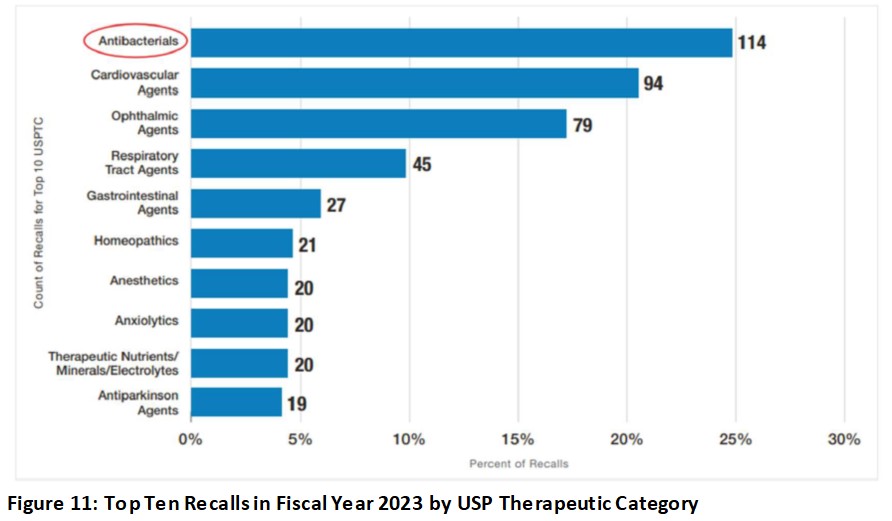 Despite having regulations and many published drug guidance documents to help bio/pharmaceutical companies aligned with FDA’s current thinking on various GMP topics, GMP deficiencies continue to evolve and repeat over the years. It is worth noting that ophthalmic agents moved up from eight in FY2022 to third, accounting for 17% of FY2023 recalls due to contamination.
Despite having regulations and many published drug guidance documents to help bio/pharmaceutical companies aligned with FDA’s current thinking on various GMP topics, GMP deficiencies continue to evolve and repeat over the years. It is worth noting that ophthalmic agents moved up from eight in FY2022 to third, accounting for 17% of FY2023 recalls due to contamination.
Contaminated eye drops could cause blindness and a warning letter was issued in October 2023 to an Indian drug manufacturer. As a result, FDA issued a draft guidance, Quality Considerations for Topical Opthalmic Drug Products in December 2023.
If US consumers face drug shortages continuously year after year despite high quality standard requirements from USFDA, what about the rest of the world? Recently, CGMP issues discovered in an India drug manufacturer led to major cancer drug shortages in 2023 where the FDA acted untraditionally allowing unapproved cancer drugs from China to be made available to US patients.
Just like checks and balances where FDA scrutinizes companies for not conducting adequate investigations, FDA was also faced with investigations and questions by US Congress (FINAL-Letter-to-Commissioner-Califf-re-Chemotherapy-Drug-Shortage.pdf (senate.gov)) when they allowed unapproved drugs into the US. This also prompted resumption of announced inspections in foreign companies (12_13_23_Letter_to_FDA_re_foreign_drug_inspections_follow_up).
For CGMP matters, the FDA uses US Federal Food, Drug, & Cosmetic Act, 21 CFR 211 for finished pharmaceuticals, and ICH Q7 for active pharmaceutical ingredients. USFDA mainly charges companies with adulteration or misbranding. For this article, the focus is on adulteration or CGMP. For drug adulteration charges, the following US FD&C Act’s provisions apply:
- 501(a): Insanitary conditions, failure to conform with CGMP, etc.
- 501(b): Strength, quality, or purity differing from official compendium
- 501(c): Misrepresentation of strength, etc., where drug is unrecognized in compendium
- 501(d): Mixture with or substitution of another substance
- 501(j): Deemed adulterated if owner/operator delays, denies, refuses, or limits inspection
For finished pharmaceuticals, the top 10 commonly cited observations for FY2023 under 21 CFR 211 are shown in Table 2.
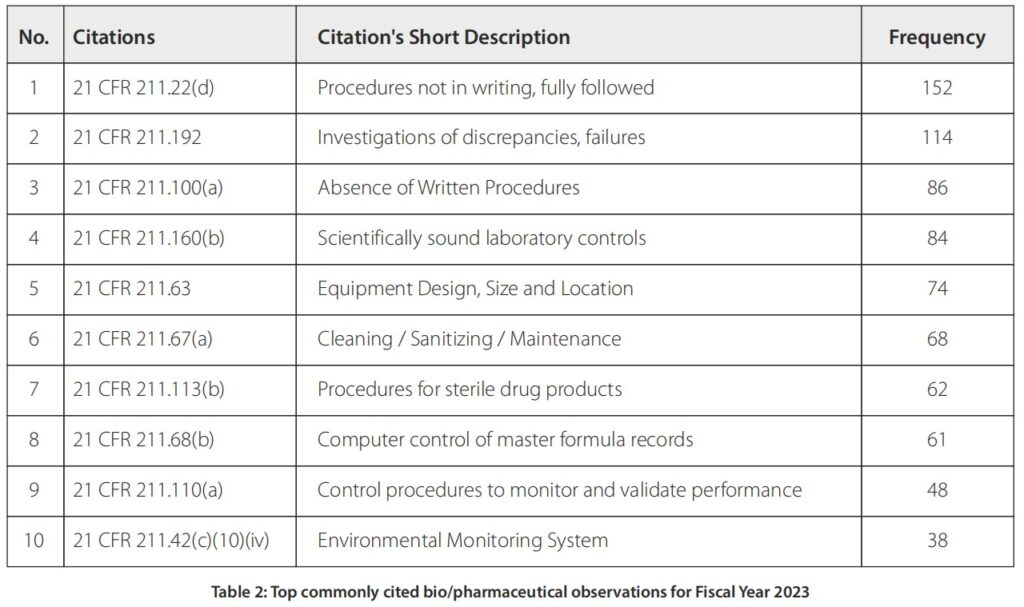 It is important to note that top ranking FDA-483 observations for finished drug products do not always translate into the same ranking when it comes to Warning Letters. For example, in 2023 the top 3 warning letter citations were 21 CFR 211.22 (responsibilities of a quality control unit), 21 CFR 211.84 (control and testing of components, containers, and closures), and 21 CFR 211.100 (written procedures, deviations). As shown in Figure 12, 21 CFR 211.84 is not listed among the top 10 commonly cited observations yet it is the 2nd top citation for warning letters in the same year of 2023. It’s important to note also that 21 CFR 211.192 was the second top citation for FDA-483 yet it is ranked as # 4 under warning letter issuance.
It is important to note that top ranking FDA-483 observations for finished drug products do not always translate into the same ranking when it comes to Warning Letters. For example, in 2023 the top 3 warning letter citations were 21 CFR 211.22 (responsibilities of a quality control unit), 21 CFR 211.84 (control and testing of components, containers, and closures), and 21 CFR 211.100 (written procedures, deviations). As shown in Figure 12, 21 CFR 211.84 is not listed among the top 10 commonly cited observations yet it is the 2nd top citation for warning letters in the same year of 2023. It’s important to note also that 21 CFR 211.192 was the second top citation for FDA-483 yet it is ranked as # 4 under warning letter issuance.
My explanation for this difference is that a warning letter is issued based on most significant issues from a firm’s FDA-483 and not what is most commonly cited among all firms in a particular year. For example, investigator(s) may cite 10 observations but warning letter may only use 5 out of 10 FDA-483 items. In addition, compliance officers may rank FDA-483 items differently than investigator(s) based on an overall risk assessment. Sometimes, FDA-483 citation may be changed by compliance as well. Therefore, one would not expect top commonly cited FDA-483 items to translate into the same ranking when it comes to warning letters.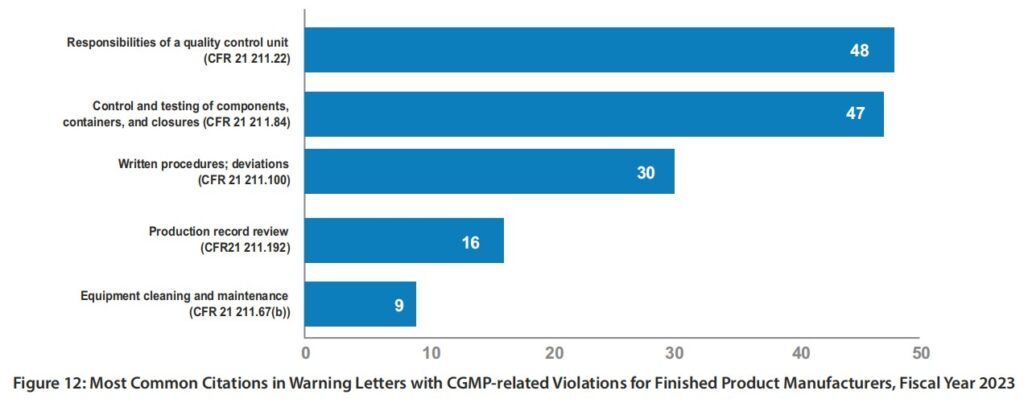
 Per Quality Management Maturity (QMM) (fda.gov), QMM looks like the umbrella (Figure 14) which to sum up is “everything quality”, if you see the implication of every word under that umbrella, in my opinion. For example, how can “business continuity” lives on if there is no product quality? What is the purpose of “Employee Ownership and Engagement” without the message to deliver high quality products? How can a company keep up with “sustainable compliance” without ensuring quality manufacturing? I hope you see the theme of “QUALITY” here.
Per Quality Management Maturity (QMM) (fda.gov), QMM looks like the umbrella (Figure 14) which to sum up is “everything quality”, if you see the implication of every word under that umbrella, in my opinion. For example, how can “business continuity” lives on if there is no product quality? What is the purpose of “Employee Ownership and Engagement” without the message to deliver high quality products? How can a company keep up with “sustainable compliance” without ensuring quality manufacturing? I hope you see the theme of “QUALITY” here.
What have you concluded from the above statistics and information based on import alert, warning letters, drug shortages, recalls due to CGMP deficiencies, and quality management maturities encompassing quality systems, quality culture, and quality metrics?
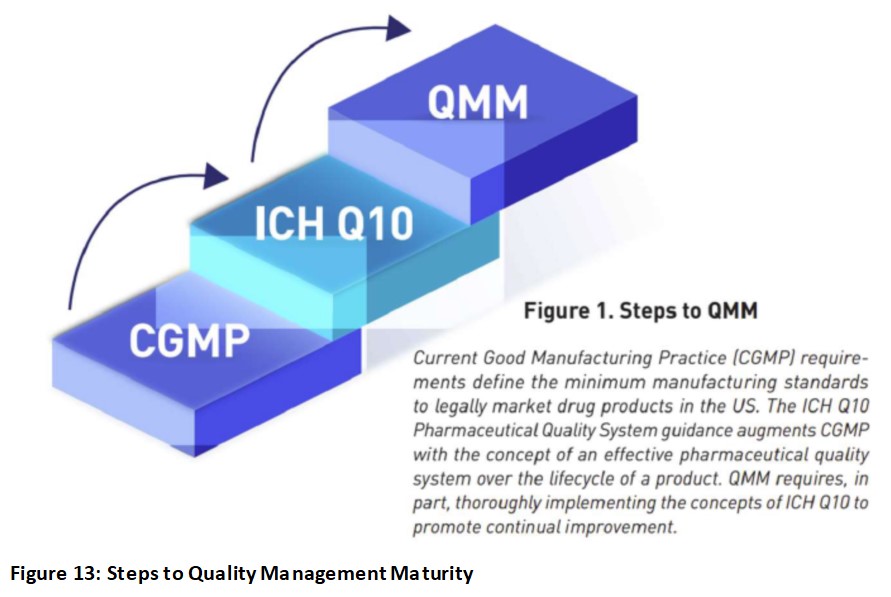
To me, it boils down to one simple word yet a challenging to achieve target: QUALITY… This is why I’m writing this article to hopefully “Providing Quality Solutions” through training, education, and development. For the bio/pharmaceutical industry, Figure 13 emphasizes the foundation of QMM which is Quality System. ICH Q10’s three main objectives are to achieve product realization, to establish and maintain a state of control, and to facilitate continual improvement.
 For me, again “Quality” is the key word, no matter how one spins this. To simplify the basic foundation of quality, I created this Figure 16 in 2009 when I was at FDA showing a different way of looking at quality systems compared to FDA (Figure 15) as described in Guidance for Industry Quality Systems Approach to Pharmaceutical CGMP Regulations in 2006.
For me, again “Quality” is the key word, no matter how one spins this. To simplify the basic foundation of quality, I created this Figure 16 in 2009 when I was at FDA showing a different way of looking at quality systems compared to FDA (Figure 15) as described in Guidance for Industry Quality Systems Approach to Pharmaceutical CGMP Regulations in 2006.
Figure 16 demonstrates CGMP quality systems beyond the six systems (Quality, Materials, Laboratory, Facilities and Equipment, Production, and Packaging and Labelling) which involves responsibilities and oversight of management and essentially anything “Quality”, which is what the photo depicts with the letter “Q” standing for the word “Quality” such as Quality culture, Quality metrics, Quality management maturity, etc. In fact, the purpose is to put every bio/pharmaceutical document which mentions or implies the word “Quality” in Figure 16.
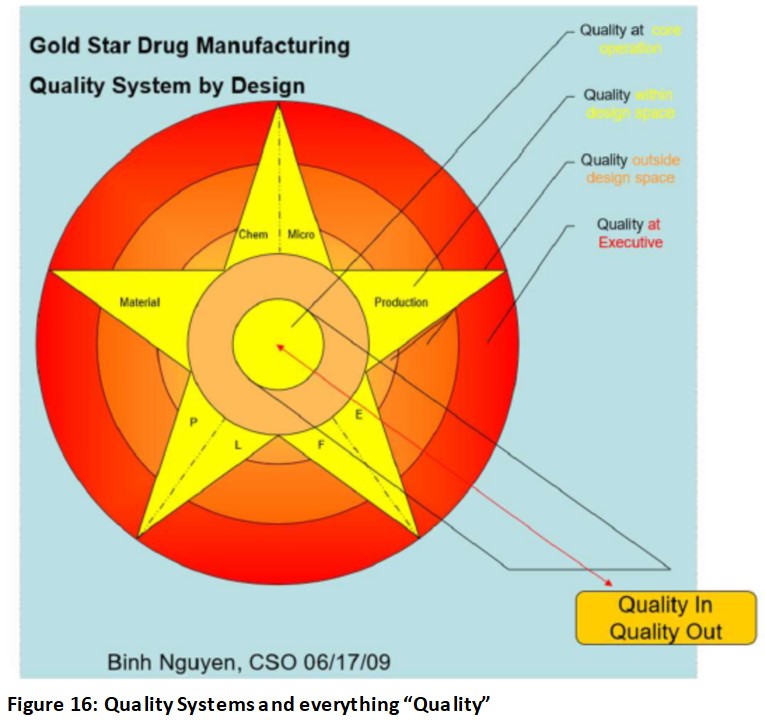 Every system (Materials, Laboratory, Facilities and Equipment, Production, and Packaging and Labelling) has to be controlled by Quality as the figure shows. Note: Quality at Executive level is highlighted in red to signify that management is ultimately responsible for the overall quality of products and manufacturing operations. A 3-D version is available but to be shown on a website or training as it is not possible here.
Every system (Materials, Laboratory, Facilities and Equipment, Production, and Packaging and Labelling) has to be controlled by Quality as the figure shows. Note: Quality at Executive level is highlighted in red to signify that management is ultimately responsible for the overall quality of products and manufacturing operations. A 3-D version is available but to be shown on a website or training as it is not possible here.
Did you know it takes about 5-10 years to train a drug FDA investigator to be proficient? And even with such long and rigorous training, not all FDA investigators are created equal (the human factor). Have you noticed why certain inspections have no FDA-483 while others have significant FDA-483 observations, yes at the same site by different FDA investigators? I had the privilege of being a Level III Pharmaceutical Inspectorate and a Level II Drug Auditor auditing Level 1 FDA drug investigators to become Level II certified FDA investigators in 2010.
Even some experienced FDA investigators did not pass FDA drug certification at the time when FDA created this certification program. This shows the importance of continuous training and certifications that the Office of Regulatory Affairs (ORA) has done to ensure that FDA investigators are efficient and thorough when conducting inspections.
At the Center for Drugs Evaluation and Research (CDER) level, not all assessors (previously referred to as reviewers) are created equal as well (the human factor) while assessing companies’ facility and/or chemistry manufacturing and control (CMC) data. This is probably one of the reasons why FDA’s Office of Pharmaceutical Quality (OPQ) announced the FY2025 Experiential Learning Site Visit Program (ELSVP) which invites pharmaceutical companies to participate in site visit proposals with OPQ in order to expose OPQ staff to drug development and manufacturing processes (Federal Register :: Office of Pharmaceutical Quality Experiential Learning Site Visit Program; Program Announcement). Similar to ORA, I believe CDER recognizes the importance of training to providing quality assessment of drug applications and/or manufacturing facilities.
So why is achieving and maintaining quality so challenging? In my view, it largely comes down to human factors, which is why it all starts with training. Making pharmaceuticals is like following a complex recipe – more steps involving human intervention increase the chances of mistakes. Take the visual inspection of sterile injectable products, for example. Even with rigorous training, medical exams, and visual inspection tests, how can you ensure consistent performance across different operators? The reality is, we’re human, and everyone has good and bad days. It only takes one bad moment to affect quality.
Even with an Acceptance Quality Limit (AQL) in place – where another inspector checks the product – we’re still taking a risk that some patients may receive a product with particulate matter. To reduce this risk, additional checks, like a robotic visual inspection, may be incorporated as a redundancy to catch human errors. However, whilst increasing the use of automated activities and controls can help improving the quality of the output, human beings cannot be replaced in full and they will continue to play an important role in the future for ensuring the manufacturing of safe and effective products.
In addition, control is an inherent and critical step of any successful process. In this sense, inspections, audits and assessments will continue to play a pivotal role in pharmaceutical manufacturing.
 What I offer to the industry is the closest opportunity to think like regulators – by training and developing auditors to operate like pseudo-FDA investigators. These auditors will be trained to deliver high-impact FDA-483 like observations, similar to the ones that lead to warning letters, injunctions, consent decrees, or voluntary facility shutdowns when necessary.
What I offer to the industry is the closest opportunity to think like regulators – by training and developing auditors to operate like pseudo-FDA investigators. These auditors will be trained to deliver high-impact FDA-483 like observations, similar to the ones that lead to warning letters, injunctions, consent decrees, or voluntary facility shutdowns when necessary.
This training is paired with an advanced inspection platform powered by artificial intelligence (AI), designed to enhance both the efficiency and quality of audits. I’ve developed global inspection software with AI capabilities, initially focused on the USFDA market but evolving to serve other regulatory regions as well.
The goal of this software is to empower auditors to conduct efficient, accurate, and thorough internal audits, preparing companies for pre-approval and surveillance inspections. This proactive approach ensures readiness for FDA inspections at all times, reducing the need to react to FDA-483 observations after the fact, and at a very high cost for the reputation of the company and its resources.
For this article, I will summarize the software for the USFDA market with the focus on bio/pharmaceutical products only. The logo (Figure 17) shows an auditor or inspector with AI and IQ (Intelligence Quotient) built in. The AI’s purpose is to empower IQ of the auditor(s). The AI is at the auditor’s fingertip to ask questions and to get immediate response(s) with references to verify AI’s generated information. This is similar to the FDA’s thinking that “trust but verify” concept when it comes to auditing.
This software is referred to as ieQipTM which stands for intelligent electronic Quality inspection platform.
Are you familiar with the frustration of inefficient or ineffective audit programs but don’t have the resources to hire high qualified consultants? Are you scared by an upcoming FDA inspection which will determine the timeliness of the launch of your new blockbuster? Are you looking for a tool to prevent negative inspection outcomes that could put the supply to clients and patients at risk?
Have a look to “ieQipTM”!!! (www.ieQip.com)
Are you equipped for FDA inspections? Yes, “ieQip”!!!
ieQipTM is designed to deliver a regulatory inspection/audit platform which incorporates many concepts from regulatory bodies around the world and structures them in a program that allows professionals to perform indepth, critical assessments of manufacturing and control processes. In addition, it “measures” the status of compliance, giving you an indication of the criticality of each of the findings identified and the probable classification of the assessment and of the overall maturity of the Quality Management System – to the eyes of the FDA investigator. This is how it works:
- Intelligence is empowered by AI to boost auditor’s intelligence. At the auditor’s fingertips, s/he can pull up the information when there are no readily available answers whether it is regulatory or scientific information.
- The Red, Yellow, and Green is like the streetlights with green means go, yellow means ready to stop, and red means stop.
- In FDA terms: red represents OAI (Official Action Indicated), yellow represents VAI (Voluntary Action Indicated), and green represents NAI (No Action Indicated).
- Note: each colourhas 3 shades indicating the shifting in good or bad direction within that colour rating.
- Remember QMM rating? This software will empower each auditor to rate what they find under a particular citation using scoring of 1-10 (Figure 18) with darker colour means the issue being audited is leaning towards worse condition and lighter colour means a shift in a better condition as described below:
- Red – dark: 1, medium: 2, light: 3
- Yellow – dark: 4, medium: 5, light: 6
- Green – dark: 7, medium: 8, light: 9
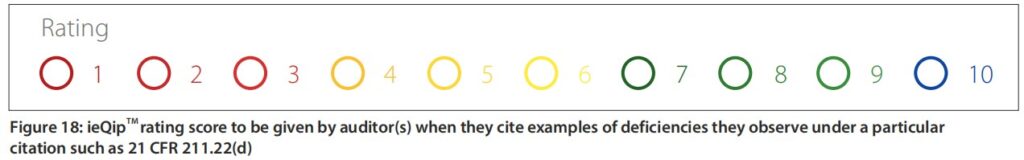 If your firm has zero FDA-483 findings, then you can have the investigator’s magnifying glass or in this case a 10. In this software, “10” rating is coloured as blue or blue sky representing that your QMM is so high that you are now flying (hopefully shipping products globally) in the blue sky and not driving dealing with streetlights.
If your firm has zero FDA-483 findings, then you can have the investigator’s magnifying glass or in this case a 10. In this software, “10” rating is coloured as blue or blue sky representing that your QMM is so high that you are now flying (hopefully shipping products globally) in the blue sky and not driving dealing with streetlights.This software’s ratings align with CDER OPQ Report on the State of Pharmaceutical Quality: FY2019, which mentions “In some instances, we use a site inspection score, on a scale of 1 to 10, which is a measure of a site’s compliance to Current Good Manufacturing Practice (CGMP) regulations.”
For me, it is impossible to rate quality culture and quality metrics because these are not written in regulations. However, I think CGMP can be rated just like how FDA classifies an inspection based on the outcome of the FDA-483. Per Figure 13 from FDA on steps to QMM, quality systems is still the foundation to get to QMM, so the better your quality systems are, the better your QMM will be in my opinion.
The key about this ieQipTM rating is that it is based on the auditor and not management who may not see specialized operations at an expert level. This rating gives the auditor the power to document and rate what they see. As you know, every auditor has a different background so they can rate based on their expertise such as chemistry, microbiology, engineer, etc.
In this software, everyone in the audit/inspection team can know what others write in real time while management can see remotely what results the entire team generate in the software which is also in real time. In other words, the rating is based on the entire team’s collaborative effort and expertise.
The worst rating would represent the worst outcome or condition of the manufacturer, and management can see the final rating and focus their effort on prioritizing and dealing with red (OAI) star rating first, and then followed by yellow (VAI) rating and green for (NAI) rating. As an example, I’m showing in Figure 19 where a red star represents the worst rating seen in an audit of a finished pharmaceutical manufacturer.
