


Dr. Sanjit Singh Lamba, Managing Partner -Trillyum Consulting and Advisory, captivated the audience at Eminences Business Media event, the cGMP Workshop 2023, with his unparalleled insights into the dynamic world of Good Manufacturing Practices (GMP). Further in response to the Pharma Machines and Technologies, he writes on the valuable insights on the most recent trends and challenges encountered by the pharmaceutical industry. But it didn’t stop there; he delved deeper, providing a comprehensive understanding of the dos and don’ts through real-world case studies.

Top 5 Trending Drug GMP issues
CGMP provides for systems that assure proper design, monitoring, and control of manufacturing processes and facilities. Adherence to the cGMP regulations assures the identity, strength, quality, and purity of drug products by requiring that manufacturers of medications adequately control manufacturing operations.
The basic requirements for GMP are listed as follows:
•Clear, written instructions and procedures
•Trained operators
•Records for manufacture and distribution
•Proper storage and distribution
•Systems for complaints and recalls.
Pharmaceuticals are subject to strict regulations due to the importance of ensuring safety, effectiveness, and the significant impact they can have on patients’ well-being. These regulations cover various aspects of pharmaceutical operations, including sales and marketing, drug price reporting, patient privacy, clinical trials, preclinical testing, and manufacturing. As a result, compliance with these regulations is a top priority for pharmaceutical companies. To ensure compliance, companies implement comprehensive good manufacturing practices and intelligence programs that involve monitoring enforcement actions by health authorities such as the FDA, including inspections, warning letters, recalls, and non-compliance reports. The pressure to comply with current good manufacturing practices (CGMP) in the pharmaceutical industry has never been higher.
In fiscal year 2022, the US FDA issued 62 warning letters and 23 import alerts to drug organizations, underscoring the importance of adhering to regulations. It is crucial to note that the FDA constantly updates its inspection priorities, focusing on emerging issues and risks. Consequently, the specific reasons behind FDA 483 observations may change over time. Pharmaceutical manufacturers can mitigate the risk of receiving such observations by fostering a culture of quality, adhering to GMP requirements, and staying up to date with the latest FDA guidance and regulations. Regular training and robust quality systems are vital for preventing compliance issues, improving product quality, and ensuring patient safety. Instead of waiting for an inspection failure, pharmaceutical companies should prioritize compliance with current good manufacturing practices (CGMP) for quality.
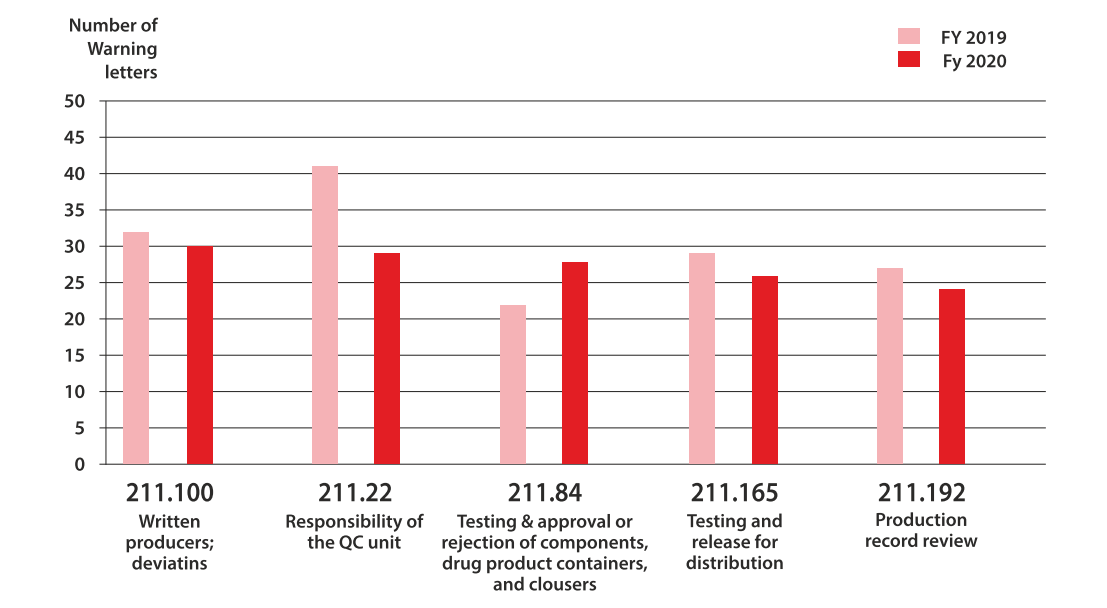
Pharmaceutical companies often face growing pressures as they approach market approval. However, it is crucial to prioritize compliance with CGMP regulations before receiving a warning. It may seem that your processes are satisfactory until an FDA inspector uncovers overlooked issues, such as unreviewed maintenance records during the absence of the lab manager.
Understanding the most common compliance issues in the pharmaceutical industry is one way to avoid receiving a 483 so you can focus on those issues instead of receiving one. The top issues in the last few years are more or less the same, but the following areas have been the most prevalent in the last five years:
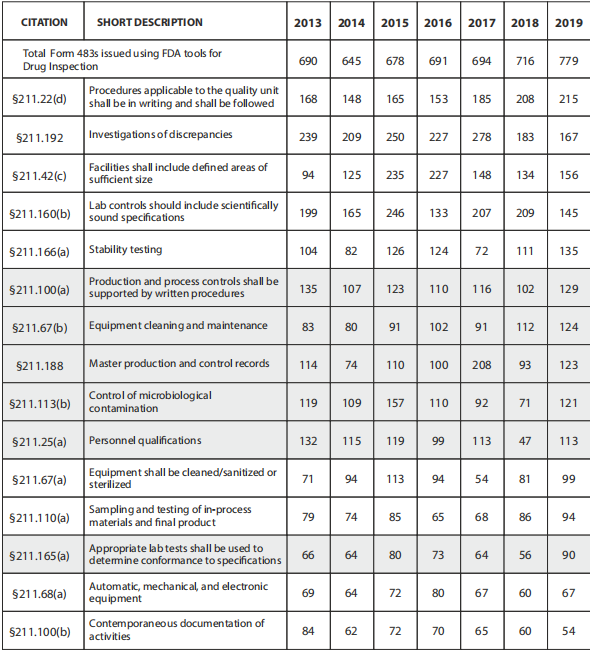
1.Absence of written procedures or failure to follow written procedures
The FDA issued 161 citations in 2022 for procedures that were not in writing or were not followed completely. Implementing, deploying, and maintaining a quality management system requires easy-to-understand procedures and work instructions.
According to the FDA Code of Federal Regulations, pharmaceutical companies must create written standard operating procedures (SOPs) for production and process control. These SOPs must include all requirements for drug identity, strength, quality, and purity. Operational and quality control units should review and approve SOPs. Whenever operations diverge from these SOPs, organizations must document compliance and create a record with a clear justification.
483 observations issued for CFR 211.100 in a recent year are often attributed to management and training issues, along with noncompliance or inadequate SOPs. Excerpts from 483 letters have included the following language:
•There are no written procedures in place at your firm
•Documented instances of non-compliance with SOPs exist
•Procedures were not approved by your firm’s quality control unit
•Documentation of SOP revisions is inadequate
•SOPs have no records of cGMP training
•The GM and Production Supervisor both stated that they were unaware of the SOP
•Despite the fact that your SOP is in English, one operator cannot read it.
With better transparency and workflows, such as the Lab Information Management system and other documentation software workflows, many of these issues can be resolved.
21 CFR 211.22(d)
“The responsibilities and procedures applicable to the quality control unit are not in writing or fully followed.”
21 CFR 211.100(a)
“There are no written procedures for production and process controls designed to assure that the drug products have the identity, strength, quality, and purity they purport or are represented to possess.”
2.Faulty production record reviews and Inadequate Investigations
21 CFR 211.192
“There is a failure to thoroughly review any unexplained discrepancy or the failure of a batch or any of its components to meet any of its specifications, whether or not the batch has been already distributed.”
In 2022, 104 pharmaceutical companies did not meet standards for investigating discrepancies and failures. Pharma QC must review and approve all drug production and control records, including packaging and labelling records, to determine compliance with standard operating procedures. The QC is responsible for conducting a thorough investigation and creating a report that includes conclusions and follow-up actions when there is a discrepancy or failure.
Red flags include:
•There is no clear understanding of the root causes
•It is common to identify root causes as “human mistakes” or similar shallow explanations
•As a result of improper categorization, the root causes are unrelated to the observations.
•Signs that someone isn’t reading what they’re signing
•Inappropriate use of investigation tools
There are a variety of ways that organizations can become non-compliant with CFR 211.192, including:
•The failure to review all logs
•Logs of downtime, cleaning, and clearance should all be reviewed
•Failures in the procedure
•It is not possible for laboratory workers to review their own work or batch records
•Procedures are not shared
•QC and operations should have a unified set of standards and SOP for batch record review to avoid confusion.
3. Failures in laboratory controls 21 CFR 211.160(b)
“Laboratory controls do not include the establishment of scientifically sound and appropriate specifications, standards, sampling plans or test procedures designed to assure that components, drug product containers, closures, in-process materials, labeling or drug products conform to appropriate standards of identity, strength, quality, and purity.”
Last year, pharmaceutical companies received 78 citations for not having scientifically sound laboratory controls. Scientifically sound laboratory controls ensure accurate, reliable lab results.
Sound lab controls comply with cGMP in the following ways:
•Scientifically sound and appropriate specifications, standards, and test procedures
•Monitoring the reliability, accuracy, precision, and performance of test procedures and instruments
•Identification and handling of test samples
An FDA laboratory inspection focuses on both lab operations and raw data to determine whether a pharma organization is compliant with cGMP. You can expect an inspector to look at:
•Lab records and logs
•SOPs
•Analytical procedures
•Raw lab data
•Lab equipment
Procedures, raw data, and management records are all necessary evidence for quality-driven operations. Establishing scientifically sound lab procedures is an enormous responsibility. In addition to instrument calibration, employee compliance with SOPs, and management investigations to determine the root cause of operations, raw data can reveal a lot about compliance and management.
4. Inadequate cleaning, sanitizing, and maintenance 21 CFR 211.67(a)
“Equipment and utensils are not cleaned, maintained, or sanitized at appropriate intervals to prevent malfunctions or contamination that would alter the safety, identity, strength, quality, or purity of the drug product.”
In 2022, the FDA slapped the wrists of 50 drug companies for poor cleaning. Essentially, if your company makes medicine, then the FDA wants to make sure that you’re working in a clean, sanitized environment to prevent malfunctions or contamination.
For cleaning and maintenance, SOPs should be followed, including:
•Cleaning responsibilities
•Schedules for maintenance and cleaning
•A detailed description of the methods to be used
•Contamination prevention for clean equipment
•Before using equipment, inspect it for cleanliness
•Keeping records of maintenance, cleaning, sanitizing, and inspection
An analysis of 483 observations for 21 CFR 211.67(a) in one recent year revealed the following reasons for noncompliance:
•On loose paper, maintenance activities have been logged as needed.
•Forty-three of approximately 55 maintenance records were not signed by the responsible employee.
•After cleaning tasks were completed, cleaning records were not reviewed and approved.
For each maintenance activity, technicians were not instructed to record data in SOPs.
There have been 483 observations within this category resulting from visible maintenance or cleaning issues. There have been reports of out-of- order equipment, holes in the ceiling of the laboratory or rust that could threaten the cleanliness of the facility. In order to prevent contamination and protect drug quality, SOPs must be reviewed frequently.
5.Control of master formula records by computer
21 CFR 211.68(b)
“Appropriate controls shall be exercised over computer or related systems to assure that changes in master production and control records or other records are instituted only by authorized personnel.”
There were 56 violations of this rule in 2022, according to the FDA.
It is possible for your critical records to be compromised when documents are not protected from unauthorized edits. Keeping track of changes and approving edits within quality management system software can prevent that and help companies comply with this regulation. It can also provide clear audit trails for FDA inspections, showing who changed what and when.
Form 483s can also be caused by:
•The degree and frequency of input/output verification is determined by the “complexity and reliability of the computer or related system” according to 21 CFR 211.68(b).
•Data backups are required by the FDA except in certain cases where automation is being used, in which case a written record and validation data are required.
Robust Root Cause Analysis Methods
During the process of conducting root cause analysis in pharmaceutical manufacturing, it is crucial to take into account the specific type of issue, available data, and potential hazards to the quality of the product and safety of the patients. To ensure a comprehensive and efficient investigation, cross-functional teams collaborate. Additionally, maintaining regulatory compliance requires documenting and tracing the analysis process and implementing corrective and preventive actions (CAPA). Robust methods of root cause analysis play a vital role in identifying the root cause of failures in pharmaceutical manufacturing and implementing effective CAPAs.

Various well-established methods and tools are utilized in the pharmaceutical industry as standard methods for conducting root cause analysis.
1.Ishikawa (Fishbone) Diagram: The Ishikawa or fishbone diagram is an effective tool for identifying potential causes of issues by categorizing factors like people, equipment, materials, processes, and environment. This helps in identifying the root cause of the problem in a structured manner.
2. 5 Whys Technique: A simple yet powerful method of asking “why” several times, usually five, to understand the root cause of a problem. This approach encourages a deeper understanding and examination of problems.
3.The Failure Mode and Effects Analysis (FMEA): A systematic approach that evaluates the probability and severity of potential failures in a process. This helps in prioritizing which problems to address first and is commonly used to prevent failures proactively.
4.Fault Tree Analysis (FTA): FTA is employed to study the various factors that may contribute to a specific event of failure. This technique involves creating a tree-like diagram that depicts the logical connections among the root causes and the ultimate failure.
5.Bowtie Analysis: Similar to FTA and is employed to assess and visualize the chain of causes and effects of an event. This technique is particularly helpful in risk assessment and the implementation of effective risk controls.
6.Pareto Analysis: The Pareto principle, which states that 80% of problems arise from 20% of the root causes. Pareto Analysis helps to identify the most significant contributors, prioritize issues and enables the organization to focus on the most impactful issues.
7.Root Cause Analysis (RCA): RCA software tools are designed for analyzing the root cause of problems. These tools feature templates, workflows, and data analysis capabilities that streamline the investigation process.
8.Statistical Methods: Statistical techniques like Regression Analysis, Design of Experiments (DOE), and Hypothesis Testing can be useful in identifying root causes when relevant data is available.
9.Fault Detection and Analysis (FDA) tools: These tools use real-time data from manufacturing processes to detect and analyze deviations and anomalies. They can help identify root causes in near-real-time, allowing for immediate corrective actions.
10.Change Analysis: this tools and methodologies are employed to assess the impact of process changes on product quality and identify any potential associated issues when investigating failures related to process changes.
11.Human Error Analysis Tools: When human error is a potential root cause, Human Error Analysis tools like the Human Error Assessment and Reduction Technique (HEART) or the Systematic Human Error Reduction and Prediction Approach (SHERPA) can be applied to understand and prevent errors.
12.The Failure Reporting, Analysis, and Corrective Action System (FRACAS): A structured approach used to collect data on failures, analyse the data to identify root causes and implement corrective actions. This approach is commonly utilized in industries such as aerospace and defence and can be adapted for pharmaceutical manufacturing.
Pressure from Leadership
Leadership is a constantly evolving and dynamic journey. To thrive in today’s fast-paced and complex business environment, leaders must be willing to let go of old habits, acquire new skills, and consistently demonstrate effective leadership behaviours. Effective leadership is an ongoing process of growth and development. Leaders should embrace the idea of unlearning, learning, and implementing new skills and behaviours in order to adapt to changing circumstances and be more successful in their roles. Here are some key areas that leaders should focus on when it comes to unlearning, learning, and practicing:

Effective Communication
Effective communication is crucial for the success of any organization, as it promotes teamwork, understanding, and cohesion among team members. Enhancing communication within a team or organization is an ongoing process that requires commitment, attentiveness, and a proactive approach to addressing any communication issues that arise. By implementing these strategies and fostering a culture of effective communication, you can greatly minimize communication challenges and improve collaboration and productivity. Tackling communication issues involves adopting practices and strategies to enhance communication at all levels. Here are some steps to implement effective communication:
1.Establish Clear Communication Goals and Objectives:
•Define clear and specific communication goals for your team or organization.
•Determine what information needs to be communicated, to whom, and why it’s important.
2.Create a Communication Plan:
•Develop a structured communication plan that outlines the key messages, channels, and timelines for communication.
•Ensure that the plan addresses both internal and external communication needs.
3.Use Multiple Communication Channels:
•Recognize that different team members may prefer different communication methods (e.g., email, in- person meetings, video conferences, messaging apps).
•Utilize a mix of channels to reach a diverse audience effectively.
4.Open and Transparent Communication:
•Foster a culture of transparency where information is shared openly, and team members feel comfortable expressing their thoughts and concerns.
•Share both good news and challenges with the team.
5.Active Listening:
•Encourage active listening by providing opportunities for team members to express their opinions and ask questions.
•Listen attentively without interrupting, and ask clarifying questions to ensure understanding.
6.Clear and Concise Messages:
•Communicate messages in a clear, concise, and easily understandable manner.
•Avoid using jargon or technical terms that team members may not be familiar with.
7.Feedback Mechanisms:
•Establish feedback mechanisms, such as regular check-ins, surveys, or suggestion boxes, to gather input and opinions from team members.
•Act on feedback and communicate the actions taken in response.
8.Training and Development:
•Provide communication training and resources to team members to improve their communication skills.
•Offer workshops or coaching on effective communication techniques.
9.Regular Meetings and Updates:
•Schedule regular team meetings, one-on-one discussions, or departmental updates to keep everyone informed and engaged.
•Stick to the schedule to ensure consistency.
10.Technology Tools:
•Leverage communication and collaboration tools and software (e.g., Slack, Microsoft Teams, project management software) to facilitate efficient and real-time communication.
11.Respect Diverse Perspectives:
•Acknowledge and respect diverse perspectives, backgrounds, and communication styles within the team.
•Encourage inclusivity and ensure that all voices are heard.
12.Leadership Role Modelling:
•Leaders should set an example by demonstrating effective communication behaviours.
•Leaders should be approachable and willing to engage in open dialogue with team members.
13.Celebrate Achievements:
•Celebrate team achievements and milestones to foster a positive and motivating atmosphere.
•Use these moments to communicate the team’s progress and future goals.
14.Adapt and Evolve:
•Continuously assess the effectiveness of your communication strategies.
•Be willing to adapt and refine your communication practices based on feedback and changing needs.
Statistical Process Control
Statistical process control (SPC) is a methodology utilized in manufacturing to effectively monitor and regulate processes to ensure quality. It involves the use of statistical techniques to measure and analyse process performance, detect variations, and make data-driven decisions to maintain consistency and quality of products. SPC is especially crucial in the pharmaceutical industry, where the utmost importance is placed on product quality and patient safety. In order to apply statistical process control appropriately in pharmaceutical manufacturing, a combination of statistical expertise, effective data management, and a commitment to quality is necessary. When implemented correctly, SPC can assist pharmaceutical companies in maintaining consistent product quality, minimizing waste and rework, and ultimately safeguarding patient safety. It is imperative to have trained personnel who possess a deep understanding of SPC principles and are equipped with the ability to accurately apply them within the manufacturing environment.
Here’s how to use SPC appropriately in pharmaceutical manufacturing:
1.Identify Critical Quality Attributes (CQAs) and Critical Process Parameters (CPPs):
•Determine the critical quality attributes (CQAs) of the pharmaceutical product. These are the characteristics that must be controlled within predefined limits to ensure the product meets its intended specifications.
•CPPs are the variables in the manufacturing process that can have a significant impact on product quality.
2.Collect data:
•Establish a data collection plan to collect relevant data from the manufacturing process. This can include measurements of temperature, pressure, flow rates, pH levels, and other data.
•Use appropriate instruments and measurement techniques to ensure consistent and accurate data collection.
3.Establish a control chart:
•X-bar charts and R-charts are commonly used for continuous data, and p-charts and np-charts for discrete data.
•The control limits are calculated using historical data or statistical methods and represent the range within which the process should operate to produce consistently high-quality products.
4.Monitor the Process:
•Continually monitor the manufacturing process using the control charts. Chart data points as they are collected.
•Analyse trends, patterns, and deviations from the control limits. These can indicate variations in the process.
5.Respond to out-of-control situations:
•An out-of-control condition occurs when data points fall outside the control limits or show non- random patterns.
•Determine whether the variation is due to common causes (inherent to the process) or special causes (unusual events).
•The process can be brought back under control by making appropriate corrections, such as adjusting parameters, recalibrating equipment, or making other necessary changes.
6.Documents and Reports:
•Keep comprehensive records of all data collected, including control charts, investigation reports, and corrective actions.
•Make sure all relevant stakeholders are informed about the status of the process and any significant deviations from the expected results.
7.Continuous Improvement:
•Using the data collected over time, analyse trends and make improvements to the process. Continuous improvement is an important aspect of statistical process control.
•To minimize the occurrence of out-of-control conditions and variations, implement preventive actions.
8.Observance of regulations:
In the pharmaceutical industry, compliance with regulatory requirements, such as those set by the FDA, EMA, and other relevant authorities, is essential to maintaining product quality and safety.
Developing Specifications
Developing scientifically robust specifications for laboratory controls in the pharmaceutical industry is crucial for ensuring the quality and safety of pharmaceutical products. These specifications outline the acceptable limits and criteria that raw materials, intermediate products, and finished products must adhere to. To guide the development of such specifications, here is a systematic approach that follows a scientifically sound process.
Understand Regulatory Requirements:
Familiarize yourself with the regulatory requirements specific to your region, such as those provided by the CDSCO in India or FDA (United States) or the European Medicines Agency (EMA). Regulations often provide guidance on establishing specifications.
Identify Critical Quality Attributes (CQAs):
Determine the critical quality attributes (CQAs) of the pharmaceutical product. CQAs are the physical, chemical, biological, or microbiological characteristics that must be controlled to ensure product quality and safety. This is a crucial step in specification development.
Review Available Data:
Collect and review historical data, including data from process development, stability studies, and prior manufacturing batches. This data can provide insights into the variability and trends associated with the product and its components.
Select Analytical Methods:
Choose appropriate analytical methods to test and measure the identified CQAs. These methods should be validated and capable of providing accurate and precise results.
Establish Acceptance Criteria:
Define acceptance criteria for each CQA based on scientific principles, risk assessments, and regulatory guidelines. Acceptance criteria should consider the intended use of the product and potential patient safety concerns.
Consider Variability and Clinical Impact:
Assess the variability of CQAs and determine their clinical impact. This involves understanding how variations in CQAs can affect the safety and efficacy of the product.
Use Risk Assessment Tools:
Consider using risk assessment tools, such as Failure Mode and Effects Analysis (FMEA) or Quality Risk Management (QRM), to evaluate the impact of variations in CQAs on product quality.
Set Realistic and Achievable Limits:
Specify limits that are achievable based on the available analytical methods and technology. Avoid setting overly stringent limits that cannot be consistently met.
Account for Manufacturing Process Changes:
Anticipate potential changes in the manufacturing process and how they might impact CQAs.
Specifications should be flexible enough to accommodate process adjustments without compromising product quality.
Consider Stability Data:
Use stability data to establish shelf-life specifications for the product. Stability studies help determine how the product’s quality attributes change over time under various storage conditions.
Statistical Tools and Data Analysis:
Utilize statistical tools, such as statistical process control (SPC) charts and trend analysis, to analyze data and set appropriate specifications.
Documentation and Justification:
Document the rationale behind each specification and the scientific basis for its establishment. This documentation is crucial for regulatory compliance and transparency.
Review and Approval:
Ensure that specifications are reviewed and approved by a cross-functional team, including experts in formulation, analytical chemistry, quality assurance, and regulatory affairs.
Periodic Review and Update:
Regularly review and, if necessary, update specifications as new data becomes available or as manufacturing processes evolve.
Compliance with Regulations:
Ensure that specifications are in compliance with relevant regulations and guidelines and that they meet the expectations of regulatory authorities.
Developing scientifically sound specifications is a complex and iterative process that requires a multidisciplinary approach and collaboration between various departments within a pharmaceutical company. Continuous monitoring and improvement of specifications are essential to maintaining product quality and ensuring patient safety throughout the product’s life cycle.
Sampling Plans and Testing Methods
Sampling plans and testing methods play a crucial role in ensuring the quality control of pharmaceutical manufacturing. Problems in these areas can have significant consequences, including compromised product quality, failure to meet regulatory requirements, and potential harm to patients. The following are some common issues that arise in relation to sampling plans and testing methods in pharmaceutical manufacturing:
1.Insufficient Sampling Frequency: Sampling too infrequently may result in the failure to detect sporadic or intermittent quality issues. On the other hand, excessive sampling can lead to resource inefficiency.
2.Inconsistent Sampling Procedures: Variability in how samples are collected or handled can introduce bias or errors into the sampling process. Standardized procedures and training are crucial.
3.Small Sample Sizes: Small sample sizes may not accurately represent the variability within a batch, potentially leading to false acceptance of non- conforming products.
4.Large Sample Sizes: Conversely, excessively large sample sizes can be time-consuming and costly, especially for expensive or time-sensitive products.
5.Unrepresentative Sampling: Failure to select samples that accurately represent the entire batch can lead to misleading results. It is important to consider techniques such as stratified sampling and randomization.
6.Improper Storage and Handling: Mishandling of samples, such as improper storage conditions, can cause degradation or contamination, which can affect test results.
7.Sampling Bias: Bias in sample selection can occur if certain parts of a batch are favoured or avoided during the sampling process. This can potentially result in inaccurate assessments of product quality.
Testing Method Issues:
1. Outdated Methods: Using outdated or inappropriate testing methods can result in inaccurate results and potential non-compliance with evolving regulatory requirements.
2. Method Validation and Verification: Failure to properly validate and verify testing methods for accuracy, precision, specificity, and sensitivity can lead to unreliable results.
3. Inadequate Training: Insufficient training of laboratory personnel can cause errors during testing, misinterpretation of results, and inconsistent reporting.
4. Instrument Calibration and Maintenance: Neglecting the calibration and maintenance of analytical instruments can result in inaccurate measurements and unreliable testing.
5. Method Transfer Issues: When transferring testing methods between locations or instruments, discrepancies or variations in results can occur if not managed properly.
6. Data Integrity: Insufficient controls and procedures can lead to data integrity issues, such as incomplete or manipulated data, compromising the accuracy and integrity of test results.
7. Stability of Reagents and Standards: The stability of reagents and standards used in testing is crucial. If these materials degrade or are mishandled, it can impact the reliability of test results.
8. Interference and Cross-Contamination: Contamination or interference from other substances or samples can result in inaccurate results. Proper cleaning and handling procedures are vital.
9. Inadequate Method Transfer: Discrepancies in equipment, environment, or personnel training when transferring methods between laboratories or manufacturers can affect the reliability of the method.
10. Regulatory Compliance: Failure to keep up with evolving regulatory requirements and guidelines for testing methods can lead to non-compliance and potential regulatory issues.
In order to tackle these problems, pharmaceutical manufacturers need to invest in strong quality control systems. This includes implementing thorough sampling plans and validated testing methods. It is crucial to continuously train, calibrate, and monitor these processes to ensure their reliability and accuracy. Collaboration between quality control, manufacturing, and research and development teams is essential for resolving issues and enhancing the overall quality of the products.
Written Procedures for Production and Process Controls
As established in 21 CFR 211.100(a) by the United States Food and Drug Administration (FDA), written procedures in manufacturing and process control are crucial for maintaining the quality, safety, and consistency of pharmaceutical goods. Common faults with these documented processes might cause compliance and quality concerns. Here are some typical concerns and solutions to them:
Common Issues:
1.Outdated Procedures: Because of changes in equipment, processes, legislation, or best practises, procedures may become obsolete.
2.Incomplete or Inaccurate Procedures: Written procedures may be missing important elements, include errors, or omit vital phases.
3.Noncompliance with Current Regulations: Procedures may not be in accordance with current regulatory requirements or industry standards.
4.Failure to Manage Versions of Written Procedures: Failure to manage versions of written procedures might result in personnel utilising obsolete materials.
5.Ineffective Change Control: Changes to processes may not be effectively recorded, evaluated, or authorised, resulting in confusion and possible compliance difficulties.
6.Poorly Communicated processes: Employees may be unaware of, or lack access to, the most recent processes, limiting their ability to follow them correctly.
How to Resolve These Issues:
1.Regular Review and Update: Implement a thorough procedure review and update process. Assign responsibilities for routine evaluations and ensure that procedures adhere to the most recent requirements and best practices.
2.Document Control: Set up a document control system to keep track of different versions of written procedures. Procedures should be clearly labeled and dated to show their status.
3.SOP Training: Make certain that staff receive thorough training on standard operating procedures (SOPs) and have access to the most recent versions.
4.Change Management: Create a systematic change management process for examining, approving, and documenting procedure modifications. This procedure should include all essential parties.
5.Cross-functional collaboration: Encourage collaboration between production, quality control, and quality assurance teams to ensure that processes are correct and effective.
6.Validation and Verification: Validate and verify procedures through testing and inspection to ensure they function as intended. This is especially significant for vital procedures.
7.In accordance with Audits: Conduct frequent internal audits to ensure that documented processes are in accordance with current rules and industry standards.
8.Employee feedback: Encourage workers to submit input on the usefulness and clarity of procedures. Consider their feedback while rewriting and updating documents.
9.Training Records: Keep records of staff procedures training to ensure that everyone is utilising the most recent versions.
10.Risk Assessment: Identify and reduce possible hazards associated with procedures using risk assessment methods such as Failure Mode and Effects Analysis (FMEA).
11.Regulatory Guidance: Keep up to speed on changes in regulations and guidance papers pertaining to documented processes, and integrate adjustments as appropriate.
12.Continuous Improvement: Foster a culture of continuous improvement by utilising feedback and lessons acquired from deviations and non- conformances to improve procedures and processes.
A methodical and forward-thinking strategy is necessary to resolve problems related to written procedures in production and process control. This approach involves tackling these issues head-on by verifying the precise, current, and regulatory- compliant nature of the procedures in question. By doing so, pharmaceutical manufacturers can ensure the quality, safety, and regulatory adherence of their products.
Unlearn, Learn and Relearn
Unlearn:
1.Change Resistance: Unlearn change resistance and embrace an adaptation culture. To remain compliant and competitive, the sector must be prepared to adapt procedures, adopt new technology, and update practises.
2.Departmental Isolation and Silos: Break down departmental silos and promote cross-functional communication. Quality and compliance are the responsibility of everyone, not only those involved in regulatory affairs or quality control.
.
3.Compliance as a Barrier: Dispel the belief that compliance is a barrier to innovation. Consider compliance to be the cornerstone for innovation and product quality.
4.Overdependence on Paper-Based methods: Unlearn your dependency on paper-based documentation methods. To enhance accuracy and traceability, migrate to electronic records and data management systems.
Learn:
1.Data Analytics and Artificial Intelligence: Discover how to use data analytics and artificial intelligence (AI) to improve quality control and compliance. These technologies can detect patterns, abnormalities, and possible problems more quickly and efficiently than human techniques.
2.Risk-Based methodologies: Learn and apply risk- based quality and compliance methodologies. Concentrate resources on high-risk areas, enabling for more efficient and focused compliance activities.
3.Ongoing Improvement: Adopt continuous improvement concepts (e.g., Lean, Six Sigma) to optimize operations, decrease waste, and increase quality. Learning problem-solving strategies and process optimization methodologies is required.
4.Supply Chain Visibility: Discover how to improve supply chain visibility and traceability, assuring raw material and component integrity across the supply chain.
Relearn:
1.Regulatory Updates: Relearn and stay up to date on changes in regulatory standards. Regulations are changing, and businesses must adapt to be compliant.
2.Cybersecurity Awareness: Refresh and enhance your cybersecurity knowledge. As processes become more digital, securing data and systems from cyber- attacks is critical for compliance and product safety.
3.Patient-Cantered Approach: Remind yourself of the significance of a patient-centred approach. While adhering to legal requirements, the sector must prioritize patient safety and satisfaction.
4.Environmental Sustainability: Relearn and rethink environmental sustainability practices.Consider the environmental effect of pharmaceutical production and look for solutions to reduce it.
5.Human Factors Engineering: Recalibrate your understanding of the importance of human factors engineering in product design and production processes. Human factors can have a big influence on product quality and patient safety.
6.Quality Culture: Continually relearn the importance of a strong quality culture. Quality should not be an afterthought but integrated into every aspect of the organization’s operations.
7.Adaptive Quality Management Systems: Remind yourself of the need of adaptive quality management systems that can swiftly adjust to changes in the industry and regulatory landscape.
Because of advances in science, technology, and regulatory requirements, the pharmaceutical sector is continually developing. Organisations must be adaptive, responsive to change, and committed to a culture of learning and continuous development in order to increase quality and compliance.
Pharmaceutical firms may effectively fulfil the demands of a dynamic and tightly regulated market by rejecting outmoded practises, adopting new skills and techniques, and strengthening the principles of quality and compliance.
Enhancing the Culture of Quality
It is crucial to enhance the culture of quality in the pharmaceutical industry to guarantee the safety, effectiveness, and adherence of pharmaceutical products. Achieving a robust quality culture necessitates dedication, strong leadership, and a continuous improvement mind set. Here are the steps to enhance the culture of quality in the pharmaceutical industry:
1.Leadership commitment: Senior leaders need to actively demonstrate their dedication to quality. This commitment should be communicated consistently throughout the organization.
2.Clarify quality values and expectations: Clearly define the values and expectations regarding quality in a quality policy or mission statement. Ensure that these principles align with the overall goals and mission of the organization.
3.Education and training: Provide comprehensive education and training on quality principles and practices to all employees, from top management to frontline staff. This training should cover regulatory requirements, standard operating procedures (SOPs), and good manufacturing practices (GMPs).
4.Empower employees: Encourage employees to take ownership of quality by empowering them. Foster an environment where they feel comfortable identifying and reporting any quality issues, near misses, or opportunities for improvement. Establish a culture where employees can voice concerns and make suggestions for enhancing quality.
5.Foster cross-functional collaboration: Encourage collaboration and cooperation between different departments, such as research and development, manufacturing, and quality control. This collaborative approach ensures that quality considerations are integrated throughout the entire product lifecycle. Encourage interdisciplinary teams to address complex quality challenges.
6.Continuous improvement: Implement continuous improvement methodologies like lean or Six Sigma to identify and eliminate waste, inefficiencies, and sources of defects in processes. Actively involve employees in improvement initiatives.
7.Root cause analysis: Promote the use of root cause analysis techniques to identify the underlying causes of quality issues and prevent their recurrence. Make sure that corrective and preventive actions (CAPAs) are implemented effectively.
8.Measurement and metrics: Establish key performance indicators (KPIs) and metrics related to quality. Regularly monitor and report on these metrics to track progress. Utilize data-driven decision-making to identify trends and areas for improvement.
9.Documented procedures: Ensure that all procedures and processes are well-documented and in compliance with regulatory standards. Emphasize the importance of consistently following established procedures.
10.Auditing and inspection: Conduct regular internal audits and inspections to assess compliance with quality standards and identify areas for improvement. Be prepared for external regulatory inspections by maintaining an inspection-ready state.
11.Quality risk management: Implement quality risk management processes to assess and mitigate risks to product quality and patient safety. Utilize risk assessments to prioritize quality-related activities.
12.Open communication: Encourage open and transparent communication at all levels of the organization. Share information on quality performance and achievements. Establish regular channels for employees to provide feedback and express concerns.
13.Recognition and reward: Recognize and reward individuals and teams for their contributions to quality improvements and adherence to quality standards. Utilize positive reinforcement to promote a culture of quality.
14.Regulatory compliance: Maintain a strong focus on regulatory compliance. Stay updated with evolving regulations and ensure that processes and products meet or exceed regulatory requirements.
15.Patient-centric approach: Prioritize patient safety and well-being in all quality-related decisions and actions. Remember that the ultimate goal is to provide safe and effective pharmaceutical products to patients.
Enhancing the culture of quality is an ongoing effort that requires commitment and vigilance at all levels of the organization. It should be woven into the fabric of the company’s operations and become an integral part of the organizational identity. Over time, a strong quality culture not only ensures regulatory compliance but also fosters trust and confidence in the pharmaceutical industry and its products.


