For quality inspectors also, it’s a core area of interest to confirm its compliance during regulatory audits to ensure that no data integrity has occurred at any stage of product cycle. Also, cGMP violations, because of non-conformance to this clause, leading to warning letters and import alerts has already invited trouble and loss of business to many reputed drug makers in past few years.
If we think and plan to work with the prospectus of Quality by Design, then 21 CFR shall be considered as a mandatory part of URS by the customer while designing and ordering the equipment.
While discussing 21 CFR, some questions always arise in the minds of all of us, like:

For more understanding, let us go through some definitions first, as an introduction to the 21CFR Part 11 with regard to its origin and journey that was started from August 20, 1997.
If we go back to the background of this regulation, it was introduced in early 1990s by FDA on the request of pharma industry for replacement of paper record with electronic records and signatures.
Under section 11.1 of subpart B (Electronic Records), all controls for closed system are clearly elaborated. The components governing control are: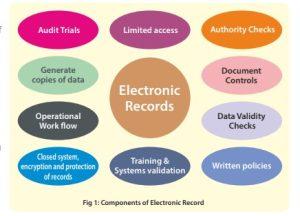
If the persons responsible for the content of electronic records also have control of system access, the system is ‘closed’. If the persons responsible for content of electronic records do not have control of system access, the system is ‘open’ (i.e., internet). Open systems require the added assurance that records are protected from point of creation to receipt.

- Non-consistency of industrial approach for software / systems development with basic requirements.
- Development function and business wing in an organisation work independently with regard to the responsibilities and management of budget.
- Business gets superseded over technical requirements because of cost implications and as a result, many critical functions are omitted by purchase as a short term savings.
- Lack of approach by industry for application of same degree of rigour and control to its management for qualification of software vendors as done in case of pharmaceutical ingredients vendor.
- Dependence and high reliability on vendor-developed and vendor-maintained software and systems regardless of what our requirements are.
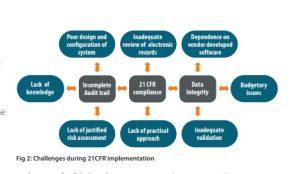
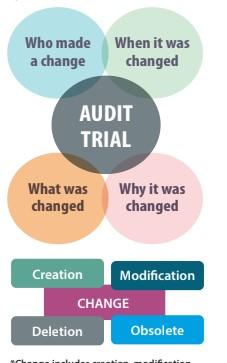
- Lack of justified and documented risk assessment” in three key areas – the need for and extent of validation, the application of audit trails, and the retention of records.
- Unlike the knowledge and capabilities that have been developed in the other
prominent industries like medical device regarding the understanding and management of risk, the pharmaceutical industry generally has much more limited experience in the formalized application of hazard control methodologies.
 About the Author
About the Author
Navdeep Singh Kathuria is General Manager – Packing, Aurobindo Pharma Ltd. He is working on implementing lean methodology along with automation of process equipment. Started his career with Tablet manufacturing, his additional area of expertise is Track & Trace systems and simplification of documentation on shop floor by means of replacing manual recording into the electronic form (EBMR).