

 Pradip Bhalerao, Founder and Director of Pharmadeep Turnkey Consultants & Engineers Pvt. Ltd., is a respected figure in the Indian pharmaceutical project industry, with an impressive experience spanning over 35 years. He has worked on a wide range of pharmaceutical projects, progressing from middle management to the esteemed position of Project Director. His expertise covers various aspects, from OSD manufacturing to vaccines and biosimilar Products.
Pradip Bhalerao, Founder and Director of Pharmadeep Turnkey Consultants & Engineers Pvt. Ltd., is a respected figure in the Indian pharmaceutical project industry, with an impressive experience spanning over 35 years. He has worked on a wide range of pharmaceutical projects, progressing from middle management to the esteemed position of Project Director. His expertise covers various aspects, from OSD manufacturing to vaccines and biosimilar Products.
Pradip has played a crucial role in enhancing the status of the Indian pharmaceutical project industry. He has been instrumental in helping several companies establish state-of-the-art manufacturing plants. His project work extends to major Asian countries, 14 African countries, and 2 European countries, showcasing his global reach and impact.
Assisting Pradip is a competent team of employees and associates based in India and Europe, who work together to serve an impressive list of clients.
In summary, Pradip Bhalerao’s remarkable journey in the pharmaceutical projects domain has left a significant mark in the industry. In recognition of his dedication, leadership, and exceptional contributions to the field of pharmaceutical project management, he was bestowed with an honorary doctorate.
Cleanrooms, by definition, are designed to control airborne particulate and environmental conditions. Cleanrooms can be positive or negative pressure environments that sweep a specified area with HEPA-filtered air.
For a successful validation, it is of utmost importance to have an all-inclusive and well-thought-of design for the cleanroom. The essence of any successful cleanroom validation lies in adopting Quality by Design principles.
When designing a cleanroom, it’s important to take into account the amount of space its mechanical equipment takes up. In the design stages, keeping flexibility in mind is essential, as this helps with issues of expansion and modification further down the line, as well as with the addition of new equipment and tools as necessary. Here are some requirements to consider when designing a new cleanroom:
 Room Flexibility: Additionally, a flexible cleanroom design means a quicker change-over of equipment, resulting in increased productivity, reduced cost, and minimal risk of contamination in the long term. When it comes to flexibility, there are several design elements you can employ to help make a cleanroom more “future-ready”.
Room Flexibility: Additionally, a flexible cleanroom design means a quicker change-over of equipment, resulting in increased productivity, reduced cost, and minimal risk of contamination in the long term. When it comes to flexibility, there are several design elements you can employ to help make a cleanroom more “future-ready”.
Airlocks: Since there can be a lot of dust, dirt particles, harmful chemicals, and other contaminants in these kinds of protected environments, airlocks can help minimize or prevent any changes in pressure that may compromise the workflow.
Electrostatic Discharge: During the day-to-day use of a cleanroom, even something as innocuous as a person walking across the room can create turbo-electric charges, which may inadvertently affect or damage certain things present in the room.
Temperature and Humidity: These two factors are integral to the design and function of a cleanroom. Temperature control is a must-have in any cleanroom as it helps provide the correct conditions needed for materials and instruments. Meanwhile, the control of humidity is needed to assure product handling conditions, stop work surface condensation, and reduce static electricity.
Pressurization: The cleanroom should be maintained at a static pressure higher than atmospheric pressure in order to prevent infiltration by the wind for most of the applications. Additionally, the air pressure should be set up in a way that the air moves from clean to less clean areas. The exception to the positive differential pressure rule is biopharmaceutical facilities, which require the facility to be at a negative pressure when dealing with certain hazardous materials.
High-Efficiency particulate filters: Practically a necessity when it comes to cleanroom design, these filters are essential to controlling contamination since they filter incredibly small particles with a 99.999% minimum particle-collective efficiency.
The most common testing performed during a cleanroom validation is indicated in Table 1 and commonly described below:
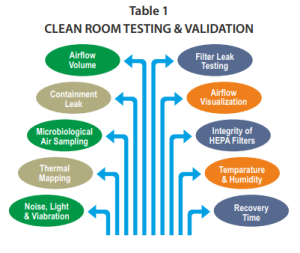 Airflow or smoke pattern: For the evaluation of this parameter, a smoke generation device is used to add a visible fume in front of the HEPA Filters or in the area in which the product shall be exposed. The distribution of smoke is observed, documented, and recorded. It should be uniform following a laminar flow pattern in the exit direction to return ducts without any major turbulence.
Airflow or smoke pattern: For the evaluation of this parameter, a smoke generation device is used to add a visible fume in front of the HEPA Filters or in the area in which the product shall be exposed. The distribution of smoke is observed, documented, and recorded. It should be uniform following a laminar flow pattern in the exit direction to return ducts without any major turbulence.
Airflow velocity and changes per hour: For this test, the area of HVAC is divided into hypothetical grids, and the air velocity is measured at each grid and then the average air velocity (V) is calculated. The area of the HEPA filter inlet (A) is calculated in feet and the total air volume (T) is then calculated by multiplying the average velocity of the air and the area of the inlet (T = A × V). After this, the volume of the room is calculated and the air changes per hour are obtained by dividing the total air change by the volume of the room.
Filter leak test: For the leak test of the HEPA filter, a velometer is placed at the front of the AHU system and the air velocity is checked. The air velocity should be within the higher limit of the HEPA filter.
Particle count: A particle counter is used to conduct the test. Particle count is taken at static conditions before the operation as well as operational working conditions. The particle count should be within the range as per the standards of particle classification, for example, ISO Class 7, etc.
Viable particle monitoring: Viable monitoring is performed on a daily basis by employing the swab test and using a nutrient agar medium for the incubation of microorganisms. The different media plates are exposed in every manufacturing section. The microorganism count should be within the range otherwise, an investigation must be initiated to evaluate the root cause, effective corrective and preventive actions
Filter integrity test: The HEPA filter integrity is tested by injecting particles of a predetermined size (0.2 um or greater) using an aerosol generator into the HEPA filters to determine if they are retaining the aerosol particles. The 100% upward flow of the aerosol must be captured into the HEPA filter. A receptor probe that detects the aerosol is used to determine if they are passing through the HEPA filter or not. Each HEPA filter must be tested and monitored periodically (e.g. annually or every six months). It is important to know if they are broken. Therefore, the amount of the aerosol detected passing through it is monitored and documented as part of the qualification. No residues or traces of aerosol must be detected after the HEPA filter to pass the acceptance criteria of the filter integrity test.
Pressure difference: It is calculated by making use of the mechanical gauges or pressure transmitters attached to the walls of the adjacent area. The pressure difference is generally kept positive from the cleanest area to the less clean area in the range from 10 and 30 mmHg pressure depending on the class of air and nature of operations.
Recovery test: The recovery of temperature and humidity conditions is checked after losing operational power conditions or doors opening. For example, the humidity and temperature are checked at the off position of the HVAC system. Then, the HVAC system is turned on to verify how much time it takes to recover the expected conditions, and the time required to stabilize the temperature and humidity is noted. Also, this test can be done, by opening the doors during some predetermined amount of time, then documenting the amount of time it takes to reach the expected environmental conditions.
Temperature and humidity uniformity test: The uniformity of temperature and humidity is monitored by employing a calibrated data logger, chart recorder, thermocouples, thermometer etc. The two parameters are monitored in a continuous manner, documented in the format, and stabilization is ensured within the specified limit.
Fresh air determination: The fresh air intake is observed at the inlet on the fresh air damper. The total air change is calculated. The intake of fresh air is divided by the total air change in the room and multiplied by 100 to obtain the percent fresh air intake on each cycle by the HVAC system in all the individual rooms.
The cleanroom validation process is a comprehensive and rigorous procedure as described in Table 2 that comprises four key stages:
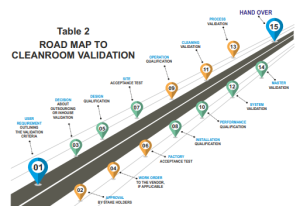 1. Design Qualification (DQ):
1. Design Qualification (DQ):
This phase ensures that the cleanroom’s design aligns with the intended requirements and meets industry standards. It encompasses evaluating the cleanroom layout, air handling systems, and filtration efficiency.
2. Installation Qualification (IQ): During IQ, the cleanroom’s installation, calibration, and configuration are meticulously tested and verified. This step confirms that the cleanroom equipment is correctly installed and performs as expected.
3. Operational Qualification (OQ): Under normal operating conditions, OQ assesses the cleanroom’s functionality. Parameters such as airflow patterns, temperature, humidity, and differential pressures are examined to validate the system’s performance.
4. Performance Qualification (PQ): PQ evaluates the cleanroom’s ability to consistently maintain the desired environmental conditions while operational. This phase is critical in demonstrating that the cleanroom complies with industry standards.
Cleanrooms are classified based on the maximum allowable airborne particle concentrations per cubic meter. This is further elaborated in Table 3.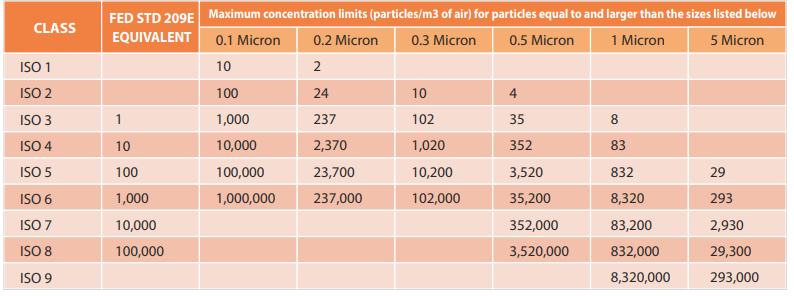
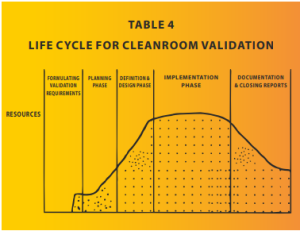
The impact of a failed cleanroom validation on the cost of a project can be significant and multi-faceted as the majority of resources are consumed by the project by the time it reaches the validation stage. This is demonstrated in Table 4. Here are some of the potential consequences:
 1. Rework and Redo Costs: When a cleanroom fails validation, it will likely require additional work, modifications, and improvements to meet the required standards. This could involve redesigning aspects of the cleanroom, purchasing new equipment, or retesting processes, all of which will incur extra expenses.
1. Rework and Redo Costs: When a cleanroom fails validation, it will likely require additional work, modifications, and improvements to meet the required standards. This could involve redesigning aspects of the cleanroom, purchasing new equipment, or retesting processes, all of which will incur extra expenses.
2. Production Delays: Failed validation means the cleanroom cannot be used for its intended purpose until the issues are resolved. This delay can lead to project timelines being extended, which in turn can increase costs due to longer operational overhead, additional labor expenses, and postponed revenue generation.
3. Compliance and Regulatory Costs: A failed validation may necessitate involvement with regulatory authorities to rectify the situation. This can lead to additional costs associated with compliance activities, potential fines, and possible penalties if the failure is severe enough to violate regulations.
4. Loss of Productivity: If the cleanroom is a critical part of the project’s operations, its failure can result in reduced productivity and efficiency. This can impact the overall project output, leading to potential losses in revenue or increased expenses to compensate for the reduced productivity.
5. Reputation and Customer Confidence: Project stakeholders, investors, and customers may lose confidence in the project if it experiences validation failures. This loss of trust can have long-term effects, affecting future project funding, partnerships, and customer relationships, which can indirectly impact the overall project’s success and financial viability.
6. Resource Allocation: Addressing cleanroom validation issues may require diverting resources (financial, human, and time) away from other project aspects. This reallocation can disrupt budgeting plans and overall resource management, potentially leading to increased costs in other areas of the project.
Overall, a failed validation of a cleanroom can have far-reaching implications, affecting not only the immediate costs of resolving the issues but also impacting the project’s overall financial performance, reputation, and success. It underscores the importance of thorough planning, adherence to Quality by Design principles, and rigorous validation procedures to mitigate the risk of failure and its associated costs.
Changing validation methods to economize on the cost of validation can lead to potential cost savings in several ways:
1. Reduced Testing Requirements: By adopting alternative validation methods, certain expensive or time-consuming tests may be replaced with more efficient and cost-effective ones. This can lead to savings in terms of labor, equipment, and materials required for testing.
2. Resource Optimization: Some validation methods might require specialized equipment or highly skilled personnel. Switching to more economical methods that utilize existing resources or less specialized personnel can result in cost savings.
3. Faster Validation Process: Certain validation methods can expedite the validation process, leading to shorter timelines and reduced operational overhead. Faster validation means quicker integration of the cleanroom into the project, which can result in cost savings.
4. Simplified Documentation: Some validation methods may require less extensive documentation compared to others. Simplified documentation can save time and effort, reducing administrative costs.
5. Risk-Based Approach: Implementing a risk-based approach to validation allows focusing on critical aspects while potentially omitting non-critical ones. This targeted approach can lead to cost savings by allocating resources where they are most needed.
6. Standardization: Adopting standardized validation methods can be cost-effective as it allows leveraging existing knowledge, templates, and best practices, reducing the need for custom solutions.
7. Regulatory Compliance Efficiency: Certain validation methods might align better with regulatory requirements, leading to smoother compliance processes and fewer rework costs due to non-compliance issues.
8. Training Costs: If the new validation methods are more straightforward and require less specialized training, the costs associated with training personnel can be reduced.
Table 5 elaborates on the relationship between the costs of change in validation criteria v/s possible cost reductions in the validation process. It is important to note that while cost-saving is a valid consideration, any changes to validation methods should not compromise the integrity and quality of the validation process.
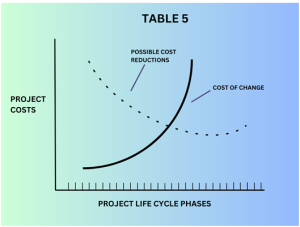 Validation is critical for ensuring the cleanroom’s functionality, safety, and compliance with industry standards and regulations.Validation is critical for ensuring the cleanroom’s functionality, safety, and compliance with industry standards and regulations. Therefore, any cost-saving measures should be thoroughly evaluated and approved by relevant stakeholders, ensuring that they do not jeopardize the reliability and effectiveness of the cleanroom.
Validation is critical for ensuring the cleanroom’s functionality, safety, and compliance with industry standards and regulations.Validation is critical for ensuring the cleanroom’s functionality, safety, and compliance with industry standards and regulations. Therefore, any cost-saving measures should be thoroughly evaluated and approved by relevant stakeholders, ensuring that they do not jeopardize the reliability and effectiveness of the cleanroom.
Additionally, the potential cost savings should be balanced against the risk of potential failures or non-compliance issues that might arise from changes in validation methods. Proper risk assessment and validation expertise are essential in making informed decisions that lead to both cost efficiency and maintaining high standards of validation.
 Introduction: This technical case study delves into the cleanroom validation process carried out at a cutting-edge nanotechnology research facility. The facility specializes in the development of nanoscale materials and devices, where even the tiniest contamination can significantly impact experimental outcomes. The stringent cleanroom validation aimed to maintain ISO 14644-1 Class 1 cleanliness standards, allowing a maximum of only 10 particles of 0.1 μm or larger per cubic meter.
Introduction: This technical case study delves into the cleanroom validation process carried out at a cutting-edge nanotechnology research facility. The facility specializes in the development of nanoscale materials and devices, where even the tiniest contamination can significantly impact experimental outcomes. The stringent cleanroom validation aimed to maintain ISO 14644-1 Class 1 cleanliness standards, allowing a maximum of only 10 particles of 0.1 μm or larger per cubic meter.
Background: The nanotechnology research facility had invested substantial resources in designing and constructing a state-of-the-art cleanroom to ensure an ultra-clean environment for conducting sensitive experiments. Given the facility’s focus on nanoscale research, ensuring the highest level of cleanliness and strict compliance with regulatory standards was crucial to the success of their research endeavors.
Validation Process
1. Design Qualification (DQ): The cleanroom validation process commenced with a design qualification phase. A multidisciplinary team of experts, including engineers, physicists, and validation specialists, thoroughly reviewed the cleanroom’s architectural plans and equipment layout. Computational fluid dynamics (CFD) simulations were employed to assess airflow patterns, particle dispersion, and identify potential contamination risks.
2. Installation Qualification (IQ): Following the construction phase, the installation qualification stage focused on rigorous testing and calibration of cleanroom equipment. The high-efficiency particulate air (HEPA) and ultra-low penetration air (ULPA) filters were subjected to challenging performance tests to verify their effectiveness in removing particles of specified sizes. Precision instruments, such as laser particle counters and differential pressure sensors, were calibrated to ensure accurate data collection.
3. Operational Qualification (OQ): The operational qualification phase involved comprehensive testing under various operating conditions. The facility’s HVAC system underwent extensive verification to ensure optimal airflow velocities, temperature uniformity, and humidity control. Pressure differentials between different cleanroom areas were carefully measured and validated to prevent cross-contamination.
4. Performance Qualification (PQ): The final stage of cleanroom validation, performance qualification, focused on the facility’s day-to-day operations. Over an extended period, the cleanroom environment was continuously monitored, and data from particle counters, air samplers, and microbial tests were analyzed. The cleanroom’s ability to consistently maintain ISO Class 1 cleanliness standards was closely scrutinized to ensure the success of nanoscale experiments.
Challenges Faced
The nanotechnology research facility encountered several complex challenges during the cleanroom validation process:
1. Particle Control at Nanoscale: Achieving ISO Class 1 cleanliness standards required rigorous control over nanoscale particles, which demanded specialized filtration and cleaning techniques.
2. Instrument Calibration: Calibrating particle counters and other monitoring instruments at nanoscale accuracy levels posed significant technical difficulties.
3. Microbial Contamination Control: Preventing microbial contamination at the nanoscale demanded stringent sterilization protocols and continuous monitoring.
Solutions Implemented
To overcome these technical challenges, the facility implemented the following solutions:
1. Advanced Filtration Technology: Cutting-edge ULPA and nano-filtration systems were employed to achieve maximum particle removal efficiency at the nanoscale.
2. Nanoscale Calibration Standards: The facility collaborated with calibration laboratories to develop nanoscale calibration standards for precise instrument calibration.
3. Sterile Protocol Implementation: Rigorous aseptic techniques, automated sterilization, and continuous microbial monitoring were instituted to minimize microbial contamination risks.
Results
The nanotechnology research facility successfully completed the cleanroom validation process, achieving ISO 14644-1 Class 1 cleanliness standards. The facility’s cleanroom demonstrated exceptional performance, maintaining ultra-clean conditions and meeting the stringent requirements for nanoscale research.




