



Concepts on Sterility Testing Isolators
The earliest uses of aseptic isolators were for sterility testing. Sterility test isolators make up most of the aseptic isolators in use and are available in many different sizes and configurations. Sterility test isolators do not need to be installed in a classified area. No formal requirement exists for a Grade D environment, but the area should be controlled to allow only trained personnel. The room should also have temperature and humidity control. Steam sterilizers used to prepare media for sterility testing were interfaced with isolators to keep the entire sterility test process under isolator conditions.
Pharmaceutical Isolator Types
Additional uses for aseptic isolators include component transfers, charging of sterile powders, and interface isolators for filling machines, depyrogenation ovens, and lyophilizers.
- Dispensing Isolators
- Sterility Testing Isolators.
- Transfer Isolators
- Sub-division and Weighing Isolators
- Cell Therapy – Cell Culture Isolators
- R&D Isolators and Class III Biosafety Cabinets
- Reactor and Charging Isolators
- Off-Loading Isolators
- Cytotoxic Compounding Containment Units
- Micronizing/Milling & Blending Isolators
Basic Working Principle/Design of Isolator:
Aseptic isolators should be free of microorganisms out of the environment and therefore need to operate under positive pressure air delivered through HEPA filters. However few isolators will work on negative pressure also. (Figure:1)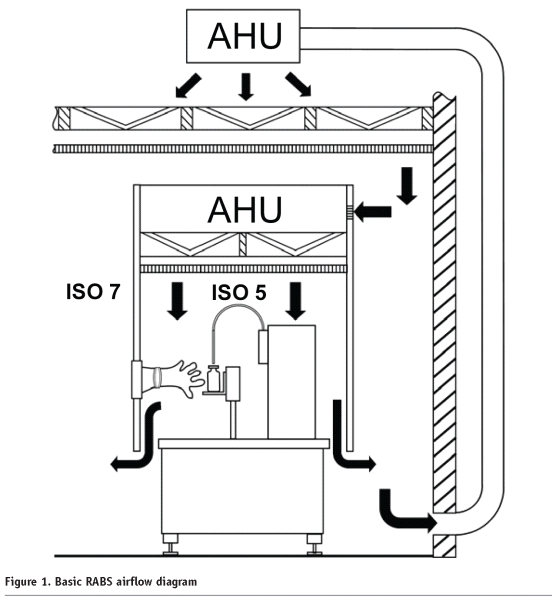
Generally this pressure is 0.25 inches of water column, but can be raised or lowered as the process requires. Isolators need to be periodically leak tested to ensure their integrity and prevent escape of the decontamination agent. Leak testing can be done by various methods including pressure decay or chemical detection.
Construction of Isolators
Aseptic isolators can be constructed using both flexible materials as well as rigid materials. Flexible wall isolators use clear plastic film (usually PVC) at a variety of thicknesses. These isolators are lighter weight, offer good visibility, and are easy to set up.
Flexible Isolators Decontamination agents are absorbed into the flexible enclosure (Figure: II) which results in long decontamination cycles while the agent “outgases” from the enclosure during aeration. Also, great care must be exercised when using sharp instruments in and around the isolator or when using cleaning agents or solvents as the flexible enclosure can be compromised.
Decontamination agents are absorbed into the flexible enclosure (Figure: II) which results in long decontamination cycles while the agent “outgases” from the enclosure during aeration. Also, great care must be exercised when using sharp instruments in and around the isolator or when using cleaning agents or solvents as the flexible enclosure can be compromised.
Rigid Wall Isolators
Rigid wall isolators are generally made from 316L stainless steel for the enclosure and laminated safety glass for viewing windows. While these isolators are heavier and take more time to install, they are more durable, do not absorb decontamination agents, which result in fast decontamination cycles, resist chemical agents, lend themselves to unidirectional airflow, and are easier to leak check than flexible wall isolators. Rigid wall isolators (Figure: III) can be used for any isolator system, especially filling machines, sterility testing, compounding and filtration, isolator systems. Where a mock-up of the design is preferred.


Steam Sterilizer Interface
Isolators are attached to exit door of pass-through sterilizer to allow direct transfer of media, supplies, etc. into the isolator system
RTP systems (double door transfers)
RTP’s are used to enter into the isolator or remove items from the isolator without breaking the “sterility” of the isolator. The RTP system is made of the two parts typically called the alpha flange and beta flange. The beta flange is rotated 60° clockwise which engages both door halves together. The operator will open the combined flanges inside the enclosure via the glove ports or half-suit. The gaskets on the flanges seal the two door halves together and the beta flange to the alpha flange (Figure: V).
RTP Docking Sequence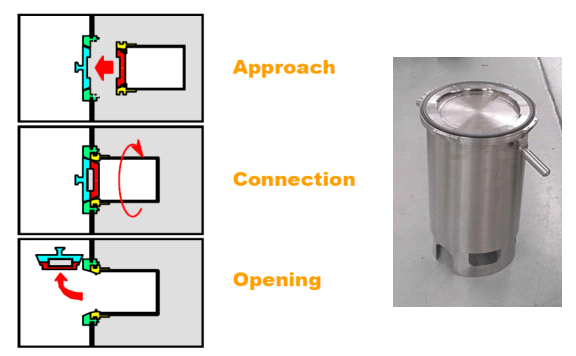
Efficacy of Cleaning
Cleaning serves to eliminate residues of the products manufactured or used. There are two approaches to cleaning:
The products are soluble in a solvent and are eliminated by dilution. In order to increase the solubility of products, surfactants and co-solvents can be added. The products are not soluble or are too hazardous to be handled as they are. A suitable chemical product is used to neutralize them or make them soluble.
The efficacy of a cleaning operation is dependent on several factors including:
- The detergent solution used
- The volume of the solution: sufficient to dissolve surface contamination but also recoverable by wiping with a suitable wipe
- The contact time which may be different from that used for a disinfectant (around 5 minutes)
- The method of application
- The number of wipes/wiping cloths used.
- Cleaning validation should be carried out on the product (active agent, excipient, etc.), the most difficult to remove of the complete procedure.
Acceptable residue limits (product manufactured, detergents, possible breakdown by-products of the product) on critical surfaces (direct or indirect contacts with the product), after cleaning, must be established and justified on the basis of a risk analysis and then validated. e.g.: Weighing, handling and formulation preparation isolators. The acceptable limits will take into account in particular the subsequent bio-decontamination, the risks of cross contamination between products, and the toxicity for maintenance operators. An analysis method specific to the residues (such as HPLC coupled with mass spectrometry, or mass spectrometry) is preferable to non-specific general methods (TOC, etc.)
The efficacy of surface sterilization (or bio-decontamination) depends on the condition of the surfaces. It should once again be stated that we are dealing here with the sterilization of clean exposed surfaces. So, the cleanliness of surfaces and their temperature are essential to ensure good surface sterilization. As a consequence, the cleaning and preparation process prior to the surface sterilization cycle must be the subject of a written procedure that is understood by the personnel involved. ‘Good practice’ in cleaning must be applied to isolators: the cleaning of isolators must be thorough and as a minimum be confirmed by visual inspection.
Leak test in Isolators
Isolators are frequently leak tested by a test known as the pressure decay test. Pressure decay may be reported in a variety of forms but the most common form is Percentage Volume Change per Hour. This volume change is actually a volume loss in positive pressure isolators and a volume gain in negative pressure Isolators.
The Percentage Volume Change per Hour is the volume of air leaked out of or into the isolator during the period of the test, expressed as a percentage of the total volume of the isolator per hour.
What guidance says?
According to ISO 10648-2, ‘The leak rate is measured at the normal operating pressure (usually about 250Pa) for checking during operational use, and 1000Pa for the acceptance test’. These test values are not generally appropriate for pharmaceutical isolators for routine testing. According to the Pharmaceutical Isolator Yellow Guide and PHSS Technical Monograph, “It is suggested that test pressure should be a minimum of (1.5 x working pressure) but may be higher (2x) depending on the design and application of the isolator”.
Industry Practice (Leak Test)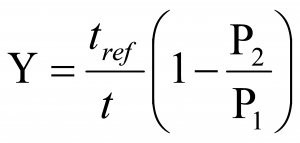 Leakage is tested using an automatic pressure loss test, during which a defined positive pressure is generated in the isolator segment in contrast to the installation room and the loss of pressure is observed for a certain time. The isolator’ tightness has to ensure a leak of air volume in 1 hour ≤ 1%. The leakage percentage is calculated as percentage of the volume of the isolator in one hour by the following formula
Leakage is tested using an automatic pressure loss test, during which a defined positive pressure is generated in the isolator segment in contrast to the installation room and the loss of pressure is observed for a certain time. The isolator’ tightness has to ensure a leak of air volume in 1 hour ≤ 1%. The leakage percentage is calculated as percentage of the volume of the isolator in one hour by the following formula
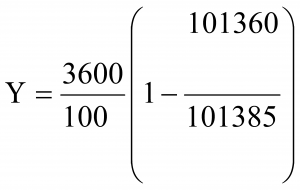
Decontamination of Isolators
Today’s isolators are decontaminated primarily with hydrogen peroxide delivered as either a gas or a condensing vapour depending on the type of generator selected. Chlorine dioxide is also used, but is not that common. Hence isolators are most commonly sanitized using hydrogen peroxide vapor (a surface disinfectant). Isolators are said to ‘disinfect’ or to ‘sanitize’ rather than ‘sterilize’ because absolute sterility cannot be demonstrated. Sanitization, in this context, describes the reduction of a number of microorganisms within the clean environment as demonstrated through the use of biological indicators in validation studies for different isolator cycles.
Generators can be portable, which can service multiple isolators or be integrated within the isolator. Cycle times depend on the volume of the isolator, materials of construction of the isolator, materials to be decontaminated within the isolator, and isolator HVAC design. To validate decontamination cycles, multiple biological indicators typically inoculated with a minimum of 106 Geobacillus stearothermophilus spores on stainless steel coupons, are placed throughout the isolator for a worst case load along with chemical indicators.
Three successive, successful cycles as a part of cycle development resulting in no remaining spores constitute a validated cycle. Aeration of the enclosure should also be validated. Typically add 20–25% to the validated exposure time to account for potential system variability. However, we must document the level of sterilant residue that will not negatively affect the process.
Validation of Isolators
Aseptic isolator validation includes Installation Qualification (IQ), Operation Qualification (OQ) of the isolator, and the Performance Qualification (PQ) of the system which includes the decontamination generator. Typical IQ checks include equipment installation, correct materials of construction, calibration status of instruments, utility connections, filter certificates, and computer software.
Typical OQ checks include verifying that set points and alarms comply with functional specifications and isolator leak test verification. Typical PQ checks include BI, D value Determination Study, Cycle Development/PQ Studies, Sterilant Penetration Study, and Residue Effects Study, Total System Challenge.
Regulatory Aspects
Human interventions represent significant risks, especially glove interventions. This is mentioned repeatedly in FDA 483’s.
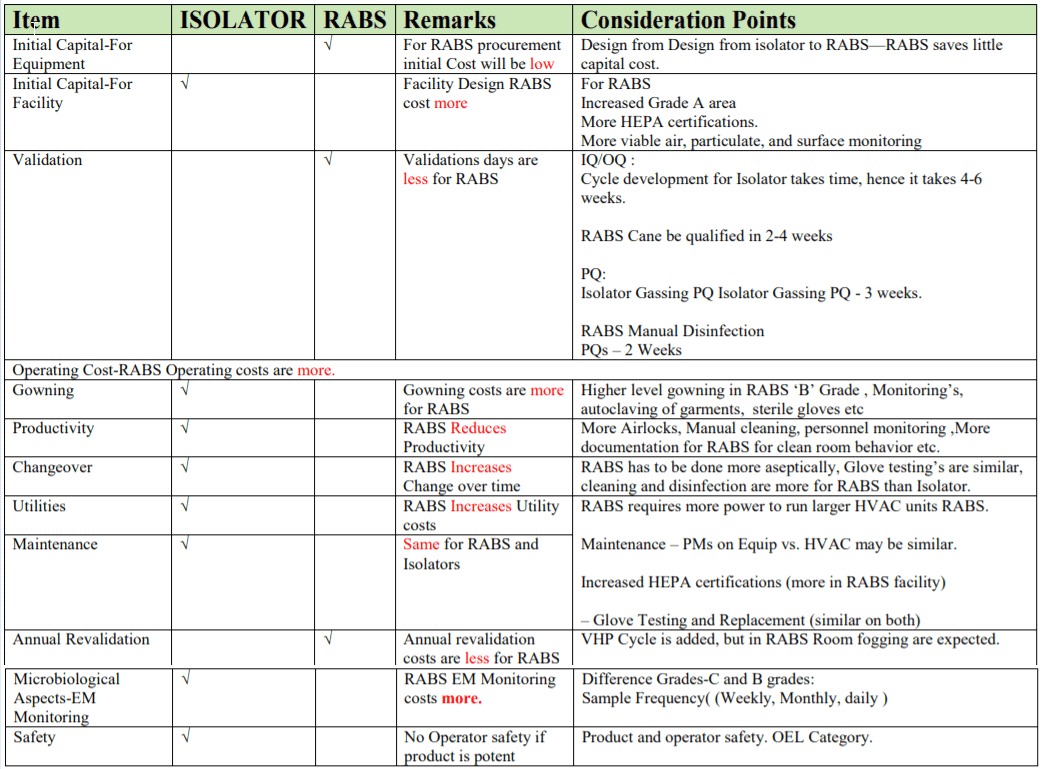
RABS:
- RABS are typically less complex and cheaper than isolators.
- Typically, no automated disinfection system.
- Surrounding room must be Grade B/ISO 7 (instead of C or D).
- Increased costs of gowning, including gowning materials and productivity losses.
- Increased environmental monitoring for surrounding ‘A’ and ‘B’ areas
RABS may be considered an attractive solution for retrofits of active solution for retrofits of existing lines, but will not replace isolation technology. Isolators and RABS will evolve as a pieces of process equipment with a defined set of functions and requirements RABS utilizes some of the advantages of isolation technology but not all of them.





